



Kürzlich wurden drei Gentherapie-Medikamente zur Vermarktung zugelassen, nämlich: (1) Am 21. Juli 2022 gab PTC Therapeutics, Inc. (NASDAQ: PTCT) bekannt, dass seine AAV-Gentherapie Upstaza™ von der Europäischen Kommission zugelassen wurde. Es handelt sich um die erste vermarktete Gentherapie, die direkt in das Gehirn injiziert wird (siehe vorherige Artikel: Ein weiterer Meilenstein in der Gentherapie | Die weltweit erste AAV-Gentherapie, die direkt in das Gehirn injiziert wird, ist zur Vermarktung zugelassen).(2) Am 17. August 2022 hat die US-amerikanische Food and Drug Administration (FDA) die Gentherapie Zynteglo (betibeglogene autotemcel, beti-cel) von Bluebird Bio zur Behandlung von Beta-Thalassämie zugelassen.Die Zulassung der Therapie in den USA ist zweifellos eine „Hilfe im Schnee“ für Bluebird Bio, das sich in einer Finanzkrise befindet.(3) Am 24. August 2022 gab BioMarin Pharmaceutical (BioMarin) bekannt, dass die Europäische Kommission die bedingte Vermarktung von ROCTAVIAN™ (Valoctocogen Roxaparvovec), einer Gentherapie für Hämophilie A, zur Behandlung von Patienten ohne Vorgeschichte von FVIII-Faktor-Inhibitoren und negativen AAV5-Antikörpern bei erwachsenen Patienten mit schwerer Hämophilie A genehmigt hat (siehe vorherigen Artikel: Heavy! BioMarins Hämophilie-A-Gentherapie zur Vermarktung zugelassen).Bisher wurden weltweit 41 Gentherapie-Medikamente zur Vermarktung zugelassen.
Gene sind die grundlegende genetische Einheit, die Merkmale steuert.Mit Ausnahme der Gene einiger Viren, die aus RNA bestehen, bestehen die Gene der meisten Organismen aus DNA.Die meisten Krankheiten des Organismus werden durch die Interaktion zwischen Genen und der Umwelt verursacht und viele Krankheiten können durch Gentherapie im Wesentlichen geheilt oder gelindert werden.Die Gentherapie gilt als Revolution in der Medizin und Pharmazie.Zu den breiten Gentherapie-Arzneimitteln gehören Arzneimittel, die auf DNA-modifizierten DNA-Arzneimitteln basieren (z. B. auf viralen Vektoren basierende In-vivo-Gentherapie-Arzneimittel, In-vitro-Gentherapie-Arzneimittel, Nacktplasmid-Arzneimittel usw.) und RNA-Arzneimittel (wie Antisense-Oligonukleotid-Arzneimittel, siRNA-Arzneimittel und mRNA-Gentherapie usw.);Eng definierte Gentherapie-Arzneimittel umfassen hauptsächlich Plasmid-DNA-Arzneimittel, Gentherapie-Arzneimittel auf Basis viraler Vektoren, Gentherapie-Arzneimittel auf Basis bakterieller Vektoren, Gene-Editing-Systeme und Zelltherapie-Arzneimittel, die in vitro genetisch verändert werden.Nach Jahren der mühsamen Entwicklung haben Gentherapie-Medikamente inspirierende klinische Ergebnisse erzielt.(ohne DNA-Impfstoffe und mRNA-Impfstoffe) wurden weltweit 41 Gentherapie-Medikamente zur Vermarktung zugelassen.Mit der Einführung von Produkten und der rasanten Entwicklung der Gentherapietechnologie steht die Gentherapie kurz vor dem Beginn einer Phase rasanter Entwicklung.
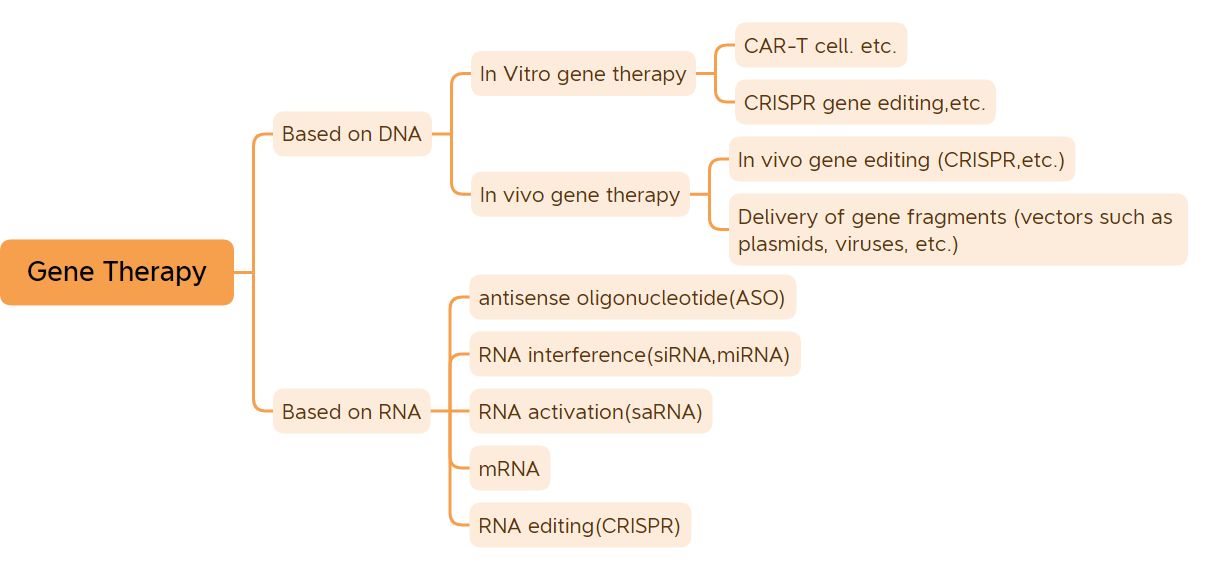
Klassifizierung der Gentherapie (Bildquelle: Biological Jingwei)
In diesem Artikel werden 41 Gentherapien aufgeführt, die zur Vermarktung zugelassen sind (ausgenommen DNA-Impfstoffe und mRNA-Impfstoffe).
1. In-vitro-Gentherapie
(1) Strimvelis
Unternehmen: Entwickelt von GlaxoSmithKline (GSK).
Zeit bis zur Markteinführung: Im Mai 2016 von der Europäischen Union zugelassen.
Indikationen: Zur Behandlung schwerer kombinierter Immunschwäche (SCID).
Anmerkungen: Der allgemeine Prozess dieser Therapie besteht darin, zunächst die eigenen hämatopoetischen Stammzellen des Patienten zu gewinnen, diese in vitro zu vermehren und zu kultivieren und dann mithilfe eines Retrovirus eine Kopie des funktionellen ADA-Gens (Adenosin-Desaminase) in seine hämatopoetischen Stammzellen einzuführen und schließlich die modifizierten hämatopoetischen Stammzellen zu übertragen.Hämatopoetische Stammzellen werden dem Körper wieder zugeführt.Klinische Ergebnisse zeigten, dass die 3-Jahres-Überlebensrate der mit Strimvelis behandelten ADA-SCID-Patienten 100 % betrug.
(2) Zalmoxis
Unternehmen: Hergestellt von MolMed, Italien.
Zeit bis zur Markteinführung: Erlangte 2016 die bedingte Marktzulassung der EU.
Indikationen: Es wird zur adjuvanten Therapie des Immunsystems von Patienten nach hämatopoetischer Stammzelltransplantation eingesetzt.
Anmerkungen: Zalmoxis ist eine allogene T-Zell-Suizidgen-Immuntherapie, die durch einen retroviralen Vektor modifiziert wird.Die Selbstmordgene 1NGFR und HSV-TK Mut2 ermöglichen es Menschen, Ganciclovir jederzeit zu verwenden, um T-Zellen abzutöten, die unerwünschte Immunreaktionen verursachen, eine weitere Verschlechterung der GVHD zu verhindern, die auftreten kann, und die Immunfunktion bei Patienten mit haploidentischer HSCT nach einer Operation wiederherzustellen. Escort.
(3) Invossa-K
Unternehmen: Entwickelt von der Firma TissueGene (KolonTissueGene).
Zeit bis zur Markteinführung: Zulassung zur Notierung in Südkorea im Juli 2017.
Indikationen: Zur Behandlung degenerativer Kniearthritis.
Anmerkungen: Invossa-K ist eine allogene Zellgentherapie, an der menschliche Chondrozyten beteiligt sind.Allogene Zellen werden in vitro genetisch verändert, und die veränderten Zellen können nach intraartikulärer Injektion den transformierenden Wachstumsfaktor β1 (TGF-β1) exprimieren und absondern.β1) und verbessert dadurch die Arthrosesymptome.Klinische Ergebnisse zeigen, dass Invossa-K Kniearthrose deutlich verbessern kann.Die Lizenz wurde 2019 von der südkoreanischen Arzneimittelbehörde widerrufen, weil der Hersteller die verwendeten Inhaltsstoffe falsch gekennzeichnet hatte.
(4) Zynteglo
Unternehmen: Entwickelt von der amerikanischen Firma bluebird bio (bluebird bio).
Zeit bis zur Markteinführung: 2019 von der Europäischen Union und im August 2022 von der FDA zugelassen.
Indikationen: Zur Behandlung der transfusionsabhängigen β-Thalassämie.
Anmerkungen: Zynteglo ist eine lentivirale In-vitro-Gentherapie, die einen lentiviralen Vektor verwendet, um eine funktionelle Kopie des normalen β-Globin-Gens (βA-T87Q-Globin-Gen) in hämatopoetische Stammzellen einzuführen, die Patienten entnommen wurden.und dann diese genetisch veränderten autologen hämatopoetischen Stammzellen dem Patienten zurück infundieren.Sobald der Patient über ein normales βA-T87Q-Globin-Gen verfügt, kann er ein normales HbAT87Q-Protein produzieren, wodurch die Notwendigkeit von Bluttransfusionen wirksam reduziert oder ganz beseitigt werden kann.Es handelt sich um eine einmalige Therapie, die lebenslange Bluttransfusionen und lebenslange Medikamente für Patienten ab 12 Jahren ersetzen soll.
(5) Skysona
Unternehmen: Entwickelt von der amerikanischen Firma bluebird bio (bluebird bio).
Zeit bis zur Markteinführung: EU-Zulassung zur Vermarktung im Juli 2021.
Indikationen: Zur Behandlung der frühen zerebralen Adrenoleukodystrophie (CALD).
Anmerkungen: Die Skysona-Gentherapie ist die einzige einmalig zugelassene Gentherapie zur Behandlung der frühen zerebralen Adrenoleukodystrophie (CALD).Skysona (Elivaldogene Autotemcel, Lenti-D) ist eine lentivirale In-vitro-Gentherapie mit hämatopoetischen Stammzellen, Lenti-D.Der allgemeine Ablauf der Therapie ist wie folgt: Dem Patienten werden autologe hämatopoetische Stammzellen entnommen, in vitro durch Lentivirus, das das menschliche ABCD1-Gen trägt, verändert und dann dem Patienten wieder infundiert.Zur Behandlung von Patienten unter 18 Jahren mit ABCD1-Genmutation und CALD.
(6) Kymriah
Unternehmen: Entwickelt von Novartis.
Zeit bis zur Markteinführung: Von der FDA im August 2017 zugelassen.
Indikationen: Behandlung der akuten lymphoblastischen Vorläufer-B-Zell-Leukämie (ALL) und des rezidivierten und refraktären DLBCL.
Anmerkungen: Kymriah ist ein lentivirales In-vitro-Gentherapie-Medikament, die weltweit erste zugelassene CAR-T-Therapie, die auf CD19 abzielt und den kostimulatorischen Faktor 4-1BB nutzt.Der Preis liegt in den USA bei 475.000 US-Dollar und in Japan bei 313.000 US-Dollar.
(7) Yescarta
Unternehmen: Entwickelt von Kite Pharma, einer Tochtergesellschaft von Gilead.
Zeit bis zur Markteinführung: Im Oktober 2017 von der FDA zugelassen.
Indikationen: Zur Behandlung von rezidiviertem oder refraktärem großzelligem B-Zell-Lymphom.
Anmerkungen: Yescarta ist eine retrovirale In-vitro-Gentherapie.Es ist die zweite zugelassene CAR-T-Therapie weltweit.Es zielt auf CD19 ab und nutzt den kostimulatorischen Faktor von CD28.Der Preis in den USA beträgt 373.000 US-Dollar.
(8) Tekartos
Unternehmen: Entwickelt von Gilead (GILD).
Zeit bis zur Markteinführung: Von der FDA im Juli 2020 zugelassen.
Indikationen: Bei rezidiviertem oder refraktärem Mantelzelllymphom.
Anmerkungen: Tecartus ist eine autologe CAR-T-Zelltherapie, die auf CD19 abzielt, und die dritte CAR-T-Therapie, die weltweit zur Vermarktung zugelassen ist.
(9) Breyanzi
Unternehmen: Entwickelt von Bristol-Myers Squibb (BMS).
Zeit bis zur Markteinführung: Von der FDA im Februar 2021 zugelassen.
Indikationen: Rezidiviertes oder refraktäres (R/R) großzelliges B-Zell-Lymphom (LBCL).
Anmerkungen: Breyanzi ist eine In-vitro-Gentherapie auf Basis des Lentivirus und die vierte CAR-T-Therapie, die weltweit zur Vermarktung zugelassen ist und auf CD19 abzielt.Die Zulassung von Breyanzi ist ein Meilenstein für Bristol-Myers Squibb auf dem Gebiet der zellulären Immuntherapie, das Bristol-Myers mit der Übernahme von Celgene im Jahr 2019 für 74 Milliarden US-Dollar erworben hat.
(10) Abecma
Unternehmen: Gemeinsam entwickelt von Bristol-Myers Squibb (BMS) und bluebird bio.
Zeit bis zur Markteinführung: Von der FDA im März 2021 zugelassen.
Indikationen: Rezidiviertes oder refraktäres multiples Myelom.
Anmerkungen: Abecma ist eine Lentivirus-basierte In-vitro-Gentherapie, die weltweit erste CAR-T-Zelltherapie gegen BCMA und die fünfte von der FDA zugelassene CAR-T-Therapie.Das Arzneimittelprinzip besteht darin, den chimären BCMA-Rezeptor auf den autologen T-Zellen des Patienten durch Lentivirus-vermittelte genetische Veränderung in vitro zu exprimieren.Vor der Infusion des Zellgen-Medikaments erhielt der Patient zur Vorbehandlung zwei Verbindungen, Cyclophosphamid und Fludarabin.Behandlung zur Entfernung unveränderter T-Zellen aus dem Patienten und anschließende Infusion der modifizierten T-Zellen zurück in den Körper des Patienten, um BCMA-exprimierende Krebszellen aufzuspüren und abzutöten.
(11) Libmeldy
Unternehmen: Entwickelt von Orchard Therapeutics.
Zeit bis zur Markteinführung: Zulassung durch die Europäische Union zur Notierung im Dezember 2020.
Indikationen: Zur Behandlung der metachromatischen Leukodystrophie (MLD).
Anmerkungen: Libmeldy ist eine Gentherapie, die auf der lentiviralen In-vitro-Genmodifikation autologer CD34+-Zellen basiert.Klinische Daten zeigen, dass eine einzelne intravenöse Infusion von Libmeldy den Verlauf einer früh einsetzenden MLD und schwerer motorischer und kognitiver Beeinträchtigungen bei unbehandelten Patienten gleichen Alters wirksam verändert.
(12) Benoda
Unternehmen: Entwickelt von WuXi Junuo.
Zeit bis zur Markteinführung: Offiziell von der NMPA im September 2021 zugelassen.
Indikationen: Behandlung von rezidiviertem oder refraktärem großzelligem B-Zell-Lymphom (r/r LBCL) bei erwachsenen Patienten nach Zweitlinien- oder systemischerer Therapie.
Anmerkungen: Benoda ist eine Anti-CD19-CAR-T-Gentherapie und auch das Kernprodukt von WuXi Junuo.Es ist das zweite in China zugelassene CAR-T-Produkt, abgesehen von rezidiviertem/refraktärem großzelligem B-Zell-Lymphom.Darüber hinaus plant WuXi Junuo auch die Entwicklung der Ruiki-Orenza-Injektion zur Behandlung verschiedener anderer Indikationen, darunter follikuläres Lymphom (FL), Mantelzell-Lymphom (MCL), chronische lymphatische Leukämie (CLL), diffuses großzelliges B-Zell-Lymphom der zweiten Wahl (DLBCL) und akute lymphatische Leukämie (ALL).
(13) CARVYKTI
Unternehmen: Das erste zugelassene Produkt von Legend Bio.
Zeit bis zur Markteinführung: Von der FDA im Februar 2022 zugelassen.
Indikationen: Behandlung von rezidiviertem oder refraktärem multiplem Myelom (R/R MM).
Anmerkungen: CARVYKTI (Ciltacabtagene Autoleucel, bezeichnet als Cilta-cel) ist eine CAR-T-Zell-Immungentherapie mit zwei Einzeldomänen-Antikörpern, die auf das B-Zell-Reifungsantigen (BCMA) abzielen.Die Daten zeigen, dass CARVYKTI eine Gesamtansprechrate von bis zu 98 % bei Patienten mit rezidiviertem oder refraktärem multiplem Myelom zeigte, die zuvor vier oder mehr Therapien erhalten hatten, darunter Proteasom-Inhibitoren, Immunmodulatoren und monoklonale Anti-CD38-Antikörper.
2. In-vivo-Gentherapie auf Basis viraler Vektoren
(1) Gendicine/Wiedergeburt
Unternehmen: Entwickelt von der Shenzhen Saibainuo Company.
Zeit bis zur Markteinführung: Zulassung zur Notierung in China im Jahr 2003.
Indikationen: Zur Behandlung von Plattenepithelkarzinomen im Kopf- und Halsbereich.
Anmerkungen: Rekombinante humane p53-Adenovirus-Injektion Gendicine/Jinshengsheng ist ein Adenovirus-Vektor-Gentherapie-Medikament mit unabhängigen geistigen Eigentumsrechten im Besitz der Shenzhen Saibainuo Company.Das Medikament besteht aus dem normalen menschlichen Tumorsuppressor-Gen p53 und künstlich modifiziertem rekombinantem Replikationsdefizit. Das humane Adenovirus Typ 5 besteht aus dem humanen Adenovirus Typ 5. Ersteres ist die Hauptstruktur des Medikaments zur Ausübung einer Antitumorwirkung, und letzteres fungiert hauptsächlich als Träger.Der Adenovirus-Vektor trägt das therapeutische Gen p53 in die Zielzelle und exprimiert das Tumorsuppressorgen p53 in der Zielzelle.Das Produkt kann verschiedene Antikrebsgene hochregulieren und die Aktivitäten verschiedener Onkogene herunterregulieren, wodurch die Antitumorwirkung des Körpers verstärkt und der Zweck der Tumortötung erreicht wird.
(2) Rigvir
Unternehmen: Entwickelt von der lettischen Firma Latima.
Zeit bis zur Markteinführung: 2004 in Lettland zugelassen.
Indikationen: Zur Behandlung von Melanomen.
Anmerkungen: Rigvir ist eine Gentherapie, die auf einem genmodifizierten ECHO-7-Enterovirus-Vektor basiert, der in Lettland, Estland, Polen, Armenien, Weißrussland und anderen Ländern eingesetzt wird und auch bei der EMA der Europäischen Union registriert wird..Klinische Fälle der letzten zehn Jahre haben gezeigt, dass das onkolytische Rigvir-Virus sicher und wirksam ist und die Überlebensrate von Melanompatienten um das 4- bis 6-fache verbessern kann.Darüber hinaus eignet sich die Therapie auch für eine Vielzahl anderer Krebsarten, darunter Darmkrebs, Bauchspeicheldrüsenkrebs, Blasenkrebs.Krebs, Nierenkrebs, Prostatakrebs, Lungenkrebs, Gebärmutterkrebs, Lymphosarkom usw.
(3) Oncorine/Ankerui
Unternehmen: Entwickelt von Shanghai Sunway Biotechnology Co., Ltd.
Zeit bis zur Markteinführung: Zulassung zur Notierung in China im Jahr 2005.
Indikationen: Behandlung von Kopf-Hals-Tumoren, Leberkrebs, Bauchspeicheldrüsenkrebs, Gebärmutterhalskrebs und anderen Krebsarten.
Anmerkungen: Oncorine ist ein Gentherapieprodukt für onkolytische Viren, das Adenovirus als Vektor verwendet.Das gewonnene onkolytische Adenovirus kann sich spezifisch in Tumoren replizieren, denen das p53-Gen fehlt oder die ein abnormales p53-Gen aufweisen, was zur Lyse von Tumorzellen führt und dadurch Tumorzellen abtötet.ohne normale Zellen zu schädigen.Klinische Ergebnisse zeigen, dass Anke Rui eine gute Sicherheit und Wirksamkeit bei einer Vielzahl bösartiger Tumoren aufweist.
(4) Glybera
Unternehmen: Entwickelt von uniQure.
Zeit bis zur Markteinführung: 2012 in Europa zugelassen.
Indikationen: Behandlung des Lipoprotein-Lipase-Mangels (LPLD) mit schweren oder wiederkehrenden Episoden einer Pankreatitis trotz streng fettreduzierter Ernährung.
Anmerkungen: Glybera (Alipogen Tiparvovec) ist ein gentherapeutisches Medikament, das auf AAV als Vektor basiert.Diese Therapie verwendet AAV als Vektor, um das therapeutische Gen LPL in Muskelzellen zu übertragen, sodass die entsprechenden Zellen eine bestimmte Menge an Lipoproteinlipase produzieren können. Es spielt eine Rolle bei der Linderung von Krankheiten und diese Therapie ist nach einer Verabreichung noch lange wirksam (die Wirkung kann viele Jahre anhalten).Das Medikament wurde 2017 von der Liste genommen, und die Gründe für die Entfernung von der Liste könnten auf zwei Faktoren zurückzuführen sein: zu hohe Preise und begrenzte Marktnachfrage.Die durchschnittlichen Kosten für eine einzelne Behandlung mit dem Medikament belaufen sich auf bis zu 1 Million US-Dollar, und bisher hat nur ein Patient es gekauft und verwendet.Obwohl die Krankenkasse 900.000 US-Dollar erstattet hat, ist es auch eine große Belastung für die Versicherung.Darüber hinaus ist die Indikation für das Medikament mit einer Inzidenzrate von etwa 1 zu 1 Million und einer hohen Rate an Fehldiagnosen zu selten.
(5) Imlygisch
Unternehmen: Entwickelt von Amgen.
Time to Market: Im Jahr 2015 wurde es zur Notierung in den Vereinigten Staaten und der Europäischen Union zugelassen.
Indikationen: Behandlung von Melanomläsionen, die durch eine Operation nicht vollständig entfernt werden können.
Anmerkungen: Imlygic ist ein gentechnisch verändertes (durch Löschung seiner ICP34.5- und ICP47-Genfragmente und Einfügen des humanen Granulozyten-Makrophagen-Kolonie-stimulierenden Faktors GM-CSF-Gens in das Virus) abgeschwächtes onkolytisches Herpes-simplex-Virus Typ 1 (HSV-1), die erste von der FDA zugelassene onkolytische Virus-Gentherapie.Die Verabreichungsmethode ist die intraläsionale Injektion.Die direkte Injektion in Melanomläsionen kann zum Aufbrechen von Tumorzellen führen und Tumorantigene und GM-CSF freisetzen, um Antitumor-Immunreaktionen zu fördern.
(6) Luxturna
Unternehmen: Entwickelt von Spark Therapeutics, einer Roche-Tochtergesellschaft.
Zeit bis zur Markteinführung: 2017 von der FDA zugelassen und 2018 für die Vermarktung in Europa zugelassen.
Indikationen: Zur Behandlung von Kindern und Erwachsenen mit Sehverlust aufgrund von Mutationen in der Doppelkopie des RPE65-Gens, aber mit ausreichender Anzahl lebensfähiger Netzhautzellen.
Anmerkungen: Luxturna ist eine AAV-basierte Gentherapie, die durch subretinale Injektion verabreicht wird.Die Gentherapie verwendet AAV2 als Träger, um eine funktionelle Kopie des normalen RPE65-Gens in die Netzhautzellen des Patienten einzuführen, sodass die entsprechenden Zellen das normale RPE65-Protein exprimieren, um den Defekt des RPE65-Proteins des Patienten zu kompensieren und so das Sehvermögen des Patienten zu verbessern.
(7) Zolgensma
Unternehmen: Entwickelt von AveXis, einer Tochtergesellschaft von Novartis.
Zeit bis zur Markteinführung: Von der FDA im Mai 2019 zugelassen.
Indikationen: Behandlung der spinalen Muskelatrophie (Spinale Muskelatrophie, SMA) bei Patienten unter 2 Jahren.
Anmerkungen: Zolgensma ist eine Gentherapie, die auf dem AAV-Vektor basiert.Dieses Medikament ist der einzige einmalige Behandlungsplan für spinale Muskelatrophie, der weltweit zur Vermarktung zugelassen ist.Seite, ist ein Meilenstein Fortschritt.Diese Gentherapie verwendet den scAAV9-Vektor, um das normale SMN1-Gen über eine intravenöse Infusion in Patienten einzuführen, wodurch normales SMN1-Protein produziert und dadurch die Funktion betroffener Zellen wie Motoneuronen verbessert wird.Im Gegensatz dazu erfordern die SMA-Medikamente Spinraza und Evrysdi eine wiederholte Gabe über einen langen Zeitraum, wobei Spinraza alle vier Monate als Wirbelsäuleninjektion verabreicht wird und Evrysdi ein tägliches orales Medikament ist.
(8) Delytact
Unternehmen: Entwickelt von Daiichi Sankyo Company Limited (TYO: 4568).
Zeit bis zur Markteinführung: Bedingte Zulassung durch das japanische Ministerium für Gesundheit, Arbeit und Soziales (MHLW) im Juni 2021.
Indikationen: Zur Behandlung von malignen Gliomen.
Anmerkungen: Delytact ist das vierte weltweit zugelassene Gentherapieprodukt für onkolytische Viren und das erste für die Behandlung von malignen Gliomen zugelassene Produkt für onkolytische Viren.Delytact ist ein gentechnisch verändertes onkolytisches Virus des Herpes-simplex-Virus Typ 1 (HSV-1), das von Dr. Todo und Kollegen entwickelt wurde.Delytact führt eine zusätzliche Deletionsmutation in das G207-Genom des HSV-1 der zweiten Generation ein, wodurch dessen selektive Replikation in Krebszellen und die Induktion von Antitumor-Immunantworten verbessert werden, während gleichzeitig ein hohes Sicherheitsprofil erhalten bleibt.Delytact ist das erste onkolytische HSV-1 der dritten Generation, das sich derzeit in der klinischen Prüfung befindet.Die Zulassung von Delytact in Japan basierte auf einer einarmigen klinischen Phase-2-Studie.Bei Patienten mit rezidivierendem Glioblastom erreichte Delytact den primären Endpunkt der Ein-Jahres-Überlebensrate, und die Ergebnisse zeigten, dass Delytact eine bessere Leistung als G207 erbrachte.Starke Replikation und höhere Antitumoraktivität.Dies ist bei soliden Tumormodellen wirksam, darunter Brust-, Prostata-, Schwannom-, Nasopharynx-, hepatozelluläre, kolorektale, bösartige Tumoren der peripheren Nervenscheide und Schilddrüsenkrebs.
(9) Upstaza
Unternehmen: Entwickelt von PTC Therapeutics, Inc. (NASDAQ: PTCT).
Zeit bis zur Markteinführung: EU-Zulassung im Juli 2022.
Indikation: Bei Mangel an aromatischer L-Aminosäure-Decarboxylase (AADC), zugelassen für die Behandlung von Patienten ab 18 Monaten.
Anmerkungen: Upstaza™ (Eladocagene Exuparvovec) ist eine In-vivo-Gentherapie unter Verwendung des Adeno-assoziierten Virus Typ 2 (AAV2) als Vektor.Der Patient ist aufgrund einer Mutation im Gen, das für das AADC-Enzym kodiert, erkrankt.AAV2 trägt ein gesundes Gen, das für das AADC-Enzym kodiert.Der therapeutische Effekt wird in Form einer genetischen Kompensation erreicht.Theoretisch ist eine Einzeldosis über einen langen Zeitraum wirksam.Es ist die erste vermarktete Gentherapie, die direkt in das Gehirn injiziert wird.Die Marktzulassung gilt für alle 27 EU-Mitgliedstaaten sowie Island, Norwegen und Liechtenstein.
(9) Roctavian
Unternehmen: Entwickelt von BioMarin Pharmaceutical (BioMarin).
Zeit bis zur Markteinführung: EU-Zulassung im August 2022.
Indikationen: Zur Behandlung erwachsener Patienten mit schwerer Hämophilie A ohne FVIII-Faktor-Hemmung in der Vorgeschichte und negativem AAV5-Antikörper.
Anmerkungen: Roctavian (Valoctocogene roxaparvovec) verwendet AAV5 als Vektor und den menschlichen leberspezifischen Promotor HLP, um die Expression des menschlichen Gerinnungsfaktors Acht (FVIII) mit gelöschter B-Domäne voranzutreiben.Die Entscheidung der Europäischen Kommission, die Vermarktung von Valoctocogen Roxaparvovec zu genehmigen, basiert auf den Gesamtdaten des klinischen Entwicklungsprogramms des Arzneimittels.Unter anderem zeigte die klinische Phase-III-Studie GENEr8-1, dass die Probanden im Vergleich zu den Daten des Jahres vor der Einschreibung nach einer einzelnen Infusion von Valoctocogen Roxaparvovec eine deutlich niedrigere jährliche Blutungsrate (ABR), eine seltenere Verwendung von rekombinanten Faktor VIII (F8)-Proteinpräparaten oder einen signifikanten Anstieg der F8-Aktivität im Körperblut aufwiesen.Nach vierwöchiger Behandlung waren der jährliche F8-Konsum und die behandlungsbedürftige ABR der Probanden um 99 % bzw. 84 % reduziert, ein statistisch signifikanter Unterschied (p < 0,001).Das Sicherheitsprofil war günstig: Bei den Probanden traten keine F8-Faktor-Hemmung, maligne oder thrombotische Nebenwirkungen auf, und es wurden keine behandlungsbedingten schwerwiegenden unerwünschten Ereignisse (SAEs) gemeldet.
3. Kleine Nukleinsäure-Medikamente
(1) Vitraven
Unternehmen: Gemeinsam entwickelt von Ionis Pharma (ehemals Isis Pharma) und Novartis.
Zeit bis zur Markteinführung: 1998 und 1999 von der FDA und der EU EMA zugelassen.
Indikationen: Zur Behandlung der Cytomegalovirus-Retinitis bei HIV-positiven Patienten.
Anmerkungen: Vitravene ist ein Antisense-Oligonukleotid-Medikament und das erste Oligonukleotid-Medikament, das weltweit zur Vermarktung zugelassen ist.Zu Beginn des Marktes war die Marktnachfrage nach Medikamenten gegen das Zytomegalievirus sehr dringend;Aufgrund der Entwicklung einer hochaktiven antiretroviralen Therapie ging die Zahl der Zytomegalievirus-Fälle dann stark zurück.Aufgrund der geringen Marktnachfrage wurde das Medikament 2002 auf den Markt gebracht und 2006 in den EU-Ländern und den USA zurückgezogen.
(2) Macugen
Unternehmen: Gemeinsam entwickelt von Pfizer und Eyetech.
Zeit bis zur Markteinführung: Zulassung zur Notierung in den Vereinigten Staaten im Jahr 2004.
Indikationen: Zur Behandlung der neovaskulären altersbedingten Makuladegeneration.
Anmerkungen: Macugen ist ein pegyliertes modifiziertes Oligonukleotid-Medikament, das auf den vaskulären endothelialen Wachstumsfaktor (VEGF165-Isoform) abzielen und an diesen binden kann und durch intravitreale Injektion verabreicht wird.
(3) Defitelio
Unternehmen: Entwickelt von Jazz.
Zeit bis zur Markteinführung: 2013 von der Europäischen Union und im März 2016 von der FDA zugelassen.
Indikationen: Zur Behandlung der Verschlusskrankheit der Lebervenen, die mit einer Nieren- oder Lungenfunktionsstörung nach einer hämatopoetischen Stammzelltransplantation einhergeht.
Anmerkungen: Defitelio ist ein Oligonukleotid-Medikament, eine Mischung aus Oligonukleotiden mit Plasmineigenschaften.Es wurde 2009 aus kommerziellen Gründen zurückgezogen.
(4) Kynamro
Unternehmen: Gemeinsam entwickelt von Ionis Pharma und Kastle.
Zeit bis zur Markteinführung: 2013 in den USA als Orphan Drug zugelassen.
Indikationen: Zur adjuvanten Behandlung der homozygoten familiären Hypercholesterinämie.
Anmerkungen: Kynamro ist ein Antisense-Oligonukleotid-Medikament, ein Antisense-Oligonukleotid, das auf menschliche Apo B-100-mRNA abzielt.Kynamro wird einmal wöchentlich in einer Dosierung von 200 mg subkutan verabreicht.
(5) Spinraza
Unternehmen: Entwickelt von Ionis Pharmaceuticals.
Zeit bis zur Markteinführung: Von der FDA im Dezember 2016 zugelassen.
Indikationen: Zur Behandlung der spinalen Muskelatrophie (SMA).
Anmerkungen: Spinraza (Nusinersen) ist ein Antisense-Oligonukleotid-Medikament.Spinraza kann das RNA-Spleißen des SMN2-Gens verändern, indem es an die Spleißstelle des SMN2-Exons 7 bindet, wodurch die Produktion von voll funktionsfähigem SMN-Protein erhöht wird.Im August 2016 übte die BIOGEN Corporation ihre Option aus, die weltweiten Rechte von Spinraza zu erwerben.Spinraza begann seine erste klinische Studie am Menschen im Jahr 2011. In nur fünf Jahren wurde es 2016 von der FDA zugelassen, was die volle Anerkennung seiner Wirksamkeit durch die FDA widerspiegelt.Das Medikament wurde im April 2019 für die Vermarktung in China zugelassen. Der gesamte Zulassungszyklus von Spinraza in China beträgt weniger als 6 Monate.Es ist zwei Jahre und zwei Monate her, seit Spinraza erstmals in den Vereinigten Staaten zugelassen wurde.Ein solches Blockbuster-Neumedikament für seltene Krankheiten im Ausland ist in Vorbereitung. Die Geschwindigkeit der Listung in China ist bereits sehr hoch.Dies ist auch auf die vom Center for Drug Evaluation am 1. November 2018 herausgegebene „Mitteilung zur Veröffentlichung der ersten Liste ausländischer neuer Medikamente, die dringend für die klinische Forschung benötigt werden“ zurückzuführen, die in die erste Charge von 40 wichtigen ausländischen neuen Medikamenten zur beschleunigten Überprüfung aufgenommen wurde und Spinraza auf Platz eins platzierte.
(6) Exondys 51
Unternehmen: Entwickelt von AVI BioPharma (später umbenannt in Sarepta Therapeutics).
Zeit bis zur Markteinführung: Im September 2016 von der FDA zugelassen.
Indikationen: Zur Behandlung der Duchenne-Muskeldystrophie (DMD) mit DMD-Genmutation im Exon-51-Skipping-Gen.
Anmerkungen: Exondys 51 ist ein Antisense-Oligonukleotid-Medikament.Das Antisense-Oligonukleotid kann an die Exon-51-Position der Prä-mRNA des DMD-Gens binden, was zur Bildung reifer mRNA führt.Durch die Exzision und dadurch teilweise Korrektur des mRNA-Leserahmens kann der Patient einige funktionelle Formen von Dystrophin synthetisieren, die kürzer als das normale Protein sind, und so die Symptome des Patienten verbessern.
(7) Tegsedi
Unternehmen: Entwickelt von Ionis Pharmaceuticals.
Zeit bis zur Markteinführung: Im Juli 2018 von der Europäischen Union zur Vermarktung zugelassen.
Indikationen: Zur Behandlung der hereditären Transthyretin-Amyloidose (hATTR).
Anmerkungen: Tegsedi ist ein Antisense-Oligonukleotid-Medikament, das auf Transthyretin-mRNA abzielt.Es ist das weltweit erste zugelassene Medikament zur Behandlung von hATTR.Die Verabreichungsmethode ist eine subkutane Injektion.Das Medikament reduziert die Produktion des ATTR-Proteins, indem es auf die mRNA von Transthyretin (ATTR) abzielt, und weist ein gutes Nutzen-Risiko-Verhältnis bei der Behandlung von ATTR auf.Weder das Krankheitsstadium noch das Vorliegen einer Kardiomyopathie waren relevant.
(8) Onpattro
Unternehmen: Gemeinsam entwickelt von Alnylam und Sanofi.
Zeit bis zur Markteinführung: Zulassung zur Notierung in den Vereinigten Staaten im Jahr 2018.
Indikationen: Zur Behandlung der hereditären Transthyretin-Amyloidose (hATTR).
Anmerkungen: Onpattro ist ein siRNA-Medikament, das auf Transthyretin-mRNA abzielt und die Produktion von ATTR-Protein in der Leber und die Ansammlung von Amyloidablagerungen in peripheren Nerven reduziert, indem es auf die mRNA von Transthyretin (ATTR) abzielt.Dadurch werden Krankheitssymptome gebessert und gelindert.
(9) Givlaari
Unternehmen: Entwickelt von Alnylam Corporation.
Zeit bis zur Markteinführung: Von der FDA im November 2019 zugelassen.
Indikationen: Zur Behandlung der akuten hepatischen Porphyrie (AHP) bei Erwachsenen.
Anmerkungen: Givlaari ist ein siRNA-Medikament, nach Onpattro das zweite zur Vermarktung zugelassene siRNA-Medikament.Das Medikament wird subkutan verabreicht und zielt auf die mRNA ab, um das ALAS1-Protein abzubauen.Eine monatliche Givlaari-Behandlung kann den ALAS1-Spiegel in der Leber deutlich und dauerhaft senken und dadurch die Werte von neurotoxischem ALA und PBG auf den normalen Bereich senken und so die Krankheitssymptome des Patienten lindern.Die Daten zeigten, dass bei mit Givlaari behandelten Patienten im Vergleich zur Placebogruppe die Zahl der Krankheitsschübe um 74 % zurückging.
(10) Vyondys53
Unternehmen: Entwickelt von Sarepta Therapeutics.
Zeit bis zur Markteinführung: Von der FDA im Dezember 2019 zugelassen.
Indikation: Zur Behandlung von DMD-Patienten mit Dystrophin-Gen-Exon-53-Splice-Mutation.
Anmerkungen: Vyondys 53 ist ein Antisense-Oligonukleotid-Medikament.Das Oligonukleotid-Medikament zielt auf den Spleißprozess des Dystrophin-mRNA-Vorläufers ab.Beim indirekten Prozess des Dystrophin-mRNA-Vorläufers wurde das externe Exon 53 teilweise herausgespleißt, d.
(11) Waylivra
Unternehmen: Entwickelt von Ionis Pharmaceuticals und seiner Tochtergesellschaft Akcea Therapeutics.
Zeit bis zur Markteinführung: Im Mai 2019 von der Europäischen Arzneimittel-Agentur (EMA) zugelassen.
Indikation: Als Zusatztherapie zu einer kontrollierten Diät bei erwachsenen Patienten mit familiärem Chylomikronämie-Syndrom (FCS).
Anmerkungen: Waylivra ist ein Antisense-Oligonukleotid-Medikament, das weltweit das erste für die Behandlung von FCS zugelassene Medikament ist.
(12) Leqvio
Unternehmen: Entwickelt von Novartis.
Zeit bis zur Markteinführung: EU-Zulassung im Dezember 2020.
Indikationen: Zur Behandlung der primären Hypercholesterinämie (heterozygote familiäre und nicht-familiäre) oder gemischten Dyslipidämie bei Erwachsenen.
Anmerkungen: Leqvio ist ein siRNA-Medikament, das auf PCSK9-mRNA abzielt.Es handelt sich um die weltweit erste siRNA-Therapie zur Senkung des Cholesterinspiegels (LDL-C).Die Verabreichungsmethode ist eine subkutane Injektion.Das Medikament wirkt durch RNA-Interferenz, um den PCSK9-Proteinspiegel zu senken, was wiederum den LDL-C-Spiegel senkt.Klinische Daten zeigen, dass Leqvio den LDL-C-Wert bei Patienten um etwa 50 % senken kann, deren LDL-C-Spiegel trotz maximal verträglicher Statindosen nicht auf die Zielwerte gesenkt werden können.
(13) Oxlumo
Unternehmen: Entwickelt von Alnylam Pharmaceuticals.
Zeit bis zur Markteinführung: EU-Zulassung im November 2020.
Indikationen: Zur Behandlung der primären Hyperoxalurie Typ 1 (PH1).
Anmerkungen: Oxlumo ist ein siRNA-Medikament, das auf die mRNA der Hydroxysäureoxidase 1 (HAO1) abzielt und subkutan verabreicht wird.Das Medikament wurde mithilfe der neuesten ESC-GalNAc-Konjugationstechnologie zur verbesserten chemischen Stabilisierung von Alnylam entwickelt, die subkutan verabreichte siRNAs mit größerer Beständigkeit und Wirksamkeit ermöglicht.Das Medikament zielt auf den Abbau oder die Hemmung der Hydroxysäureoxidase 1 (HAO1)-mRNA ab, senkt den Glykolatoxidasespiegel in der Leber und verbraucht dann das für die Oxalatproduktion erforderliche Substrat und reduziert die Oxalatproduktion, um das Fortschreiten der Krankheit zu kontrollieren und die Krankheitssymptome bei Patienten zu verbessern.
(14) Viltepso
Unternehmen: Entwickelt von NS Pharma, einer Tochtergesellschaft von Nippon Shinyaku.
Zeit bis zur Markteinführung: Von der FDA im August 2020 zugelassen.
Indikationen: Zur Behandlung der Duchenne-Muskeldystrophie (DMD) mit DMD-Genmutation im Exon-53-Skipping-Gen.
Anmerkungen: Viltepso ist ein Phosphordiamid-Morpholino-Oligonukleotid-Arzneimittel.Dieses Oligonukleotid-Medikament kann an die Exon-53-Position der Prä-mRNA des DMD-Gens binden, was zur Bildung reifer mRNA führt.Das Exon wird teilweise entfernt, wodurch der mRNA-Leserahmen teilweise korrigiert wird, was dem Patienten hilft, einige funktionelle Formen von Dystrophin zu synthetisieren, die kürzer als das normale Protein sind, wodurch sich die Symptome des Patienten verbessern.
(15) Amvuttra (Vutrisiran)
Unternehmen: Entwickelt von Alnylam Pharmaceuticals.
Zeit bis zur Markteinführung: Von der FDA im Juni 2022 zugelassen.
Indikationen: Zur Behandlung der erblichen Transthyretin-Amyloidose bei Erwachsenen mit Polyneuropathie (hATTR-PN).
Anmerkungen: Amvuttra (Vutrisiran) ist ein siRNA-Medikament, das auf Transthyretin (ATTR)-mRNA abzielt und durch subkutane Injektion verabreicht wird.Vutrisiran wurde auf der Grundlage der Enhanced Stabilization Chemistry (ESC)-GalNAc-konjugierten Verabreichungsplattform von Alnylam mit erhöhter Wirksamkeit und Stoffwechselstabilität entwickelt.Die Zulassung der Therapie basiert auf 9-Monats-Daten aus der klinischen Phase-III-Studie (HELIOS-A). Die Gesamtergebnisse zeigen, dass die Therapie die Symptome von hATTR-PN verbesserte, wobei mehr als 50 % der Patienten die Progression umkehrten oder stoppten.
4. Andere Gentherapeutika
(1) Rexin-G
Unternehmen: Entwickelt von Epeius Biotech.
Markteinführung: 2005 von der philippinischen Lebensmittel- und Arzneimittelbehörde (BFAD) zugelassen.
Indikationen: Zur Behandlung fortgeschrittener Krebserkrankungen, die gegen eine Chemotherapie resistent sind.
Anmerkungen: Rexin-G ist eine genbeladene Nanopartikel-Injektion.Es führt das mutierte Cyclin-G1-Gen über einen retroviralen Vektor in Zielzellen ein, um solide Tumoren gezielt abzutöten.Die Verabreichungsmethode ist eine intravenöse Infusion.Da es sich um ein auf den Tumor gerichtetes Medikament handelt, das aktiv nach metastasierenden Krebszellen sucht und diese zerstört, hat es eine gewisse Wirkung auf Patienten, die gegenüber anderen Krebsmedikamenten, einschließlich zielgerichteter Biologika, wirkungslos sind.
(2) Neovasculgen
Unternehmen: Entwickelt vom Institut für menschliche Stammzellen.
Zeitpunkt der Notierung: Zulassung zur Notierung in Russland am 7. Dezember 2011 und anschließende Notierung in der Ukraine im Jahr 2013.
Indikationen: Zur Behandlung peripherer arterieller Erkrankungen, einschließlich schwerer Extremitätenischämie.
Anmerkungen: Neovasculgen ist eine auf DNA-Plasmiden basierende Gentherapie, bei der das Gen für den vaskulären endothelialen Wachstumsfaktor (VEGF) 165 auf einem Plasmidrückgrat aufgebaut und Patienten infundiert wird.
(3) Collategene
Unternehmen: Gemeinsam entwickelt von der Universität Osaka und Risikokapitalfirmen.
Zeitpunkt der Listung: Vom japanischen Ministerium für Gesundheit, Arbeit und Soziales für die Listung im August 2019 genehmigt.
Indikationen: Behandlung schwerer Ischämie der unteren Extremitäten.
Anmerkungen: Collategene ist eine plasmidbasierte Gentherapie, das erste von AnGes hergestellte einheimische japanische Gentherapie-Medikament.Der Hauptbestandteil dieses Arzneimittels ist ein nacktes Plasmid, das die Gensequenz des menschlichen Hepatozyten-Wachstumsfaktors (HGF) enthält.Wenn das Medikament in die Muskeln der unteren Gliedmaßen injiziert wird, fördert der exprimierte HGF die Bildung neuer Blutgefäße um die verschlossenen Blutgefäße herum.Klinische Studien haben seine Wirksamkeit bei der Verbesserung von Geschwüren bestätigt.
ENDE
Zeitpunkt der Veröffentlichung: 10.11.2022



