



Gene sind die grundlegenden genetischen Einheiten, die Merkmale steuern.Mit Ausnahme der Gene einiger Viren, die aus RNA bestehen, bestehen die Gene der meisten Organismen aus DNA.Die meisten Krankheiten von Organismen werden durch die Interaktion zwischen Genen und der Umwelt verursacht.Durch Gentherapie können viele Krankheiten grundsätzlich geheilt oder gelindert werden.Die Gentherapie gilt als Revolution in der Medizin und Pharmazie.Zu den Gentherapie-Arzneimitteln im weitesten Sinne gehören auf DNA-modifizierte DNA-Arzneimittel (wie In-vivo-Gentherapie-Arzneimittel auf Basis viraler Vektoren, In-vitro-Gentherapie-Arzneimittel, Nacktplasmid-Arzneimittel usw.) und RNA-Arzneimittel (wie Antisense-Oligonukleotid-Arzneimittel, siRNA-Arzneimittel und mRNA-Gentherapie usw.);Im engeren Sinne umfassen Gentherapie-Medikamente hauptsächlich Plasmid-DNA-Medikamente, Gentherapie-Medikamente auf Basis viraler Vektoren, Gentherapie-Medikamente auf Basis bakterieller Vektoren, Gen-Editing-Systeme und Zelltherapie-Medikamente zur In-vitro-Genmodifikation.Nach Jahren der mühsamen Entwicklung haben Gentherapie-Medikamente ermutigende klinische Ergebnisse erzielt.(DNA-Impfstoffe und mRNA-Impfstoffe nicht eingerechnet) Derzeit sind weltweit 45 Gentherapie-Medikamente zur Vermarktung zugelassen.Insgesamt wurden in diesem Jahr 9 Gentherapien zur Vermarktung zugelassen, darunter 7 Gentherapien, die in diesem Jahr erstmals zur Vermarktung zugelassen wurden, nämlich: CARVYKTI, Amvuttra, Upstaza, Roctavian, Hemgenix, Adstiladrin und Ebvallo (Hinweis: Die anderen beiden wurden in diesem Jahr in den Vereinigten Staaten zugelassen. Die erste Charge von Gentherapien, die vermarktet werden sollen, sind: ① Zynteglo, das von der FDA für die Vermarktung in den Vereinigten Staaten zugelassen wurde August 2022 und wurde 2019 von der Europäischen Union zur Vermarktung zugelassen; .) Mit der Einführung von immer mehr Gentherapieprodukten und der rasanten Entwicklung der Gentherapietechnologie steht die Gentherapie kurz vor dem Beginn einer Phase rasanter Entwicklung.
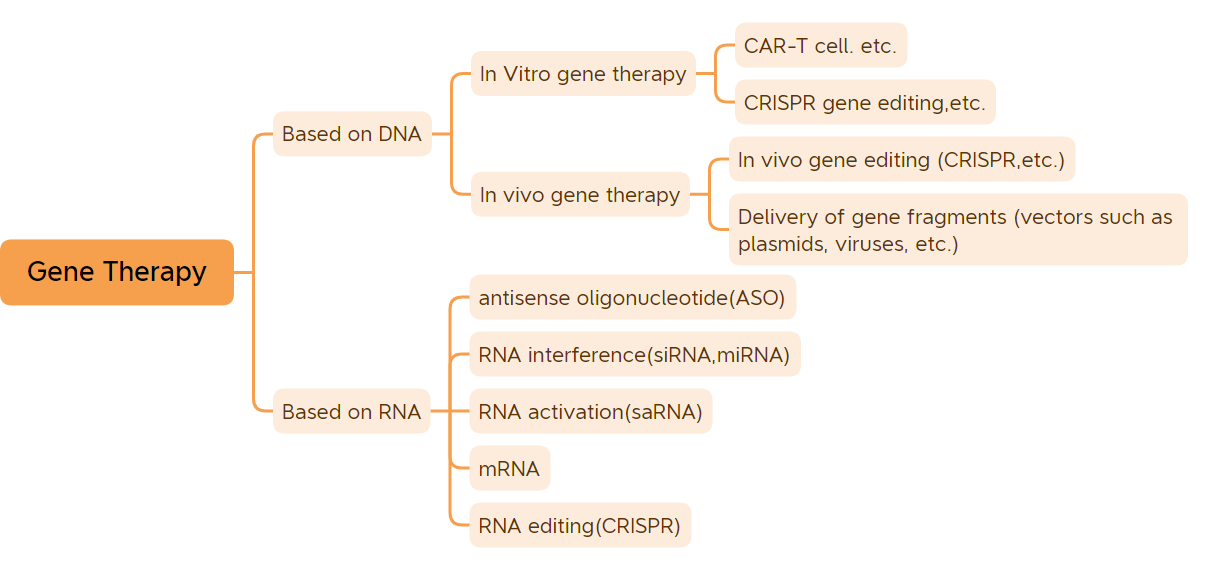
Klassifikation der Gentherapie (Bildquelle: Bio-Matrix)
In diesem Artikel werden 45 Gentherapien (ausgenommen DNA-Impfstoffe und mRNA-Impfstoffe) aufgeführt, die zur Vermarktung zugelassen sind.
1. In-vitro-Gentherapie
(1) Strimvelis
Unternehmen: Entwickelt von GlaxoSmithKline (GSK).
Zeit bis zur Markteinführung: Im Mai 2016 wurde es von der Europäischen Union zur Vermarktung zugelassen.
Indikationen: Zur Behandlung schwerer kombinierter Immunschwäche (SCID).
Anmerkungen: Der allgemeine Prozess dieser Therapie besteht darin, zunächst die eigenen hämatopoetischen Stammzellen des Patienten zu gewinnen, sie zu vermehren und in vitro zu kultivieren, dann mithilfe eines Retrovirus eine funktionelle ADA-Genkopie (Adenosin-Desaminase) in die hämatopoetischen Stammzellen einzuführen und schließlich die modifizierten hämatopoetischen Stammzellen wieder in den Körper zu injizieren.Klinische Ergebnisse zeigen, dass die 3-Jahres-Überlebensrate der mit Strimvelis behandelten ADA-SCID-Patienten 100 % beträgt.
(2) Zalmoxis
Unternehmen: Hergestellt von der italienischen MolMed Company.
Zeit bis zur Markteinführung: Erhielt 2016 die bedingte Marktzulassung der Europäischen Union.
Indikationen: Es wird zur adjuvanten Therapie des Immunsystems von Patienten nach hämatopoetischer Stammzelltransplantation eingesetzt.
Anmerkungen: Zalmoxis ist eine allogene T-Zell-Suizidgen-Immuntherapie, die durch retrovirale Vektoren modifiziert wird.Bei dieser Methode werden retrovirale Vektoren verwendet, um allogene T-Zellen genetisch zu modifizieren, sodass die genetisch veränderten T-Zellen die Suizidgene 1NGFR und HSV-TK Mut2 exprimieren. Dies ermöglicht es Menschen, jederzeit Ganciclovir-Medikamente (Ganciclovir) zu verwenden, um T-Zellen abzutöten, die unerwünschte Immunreaktionen verursachen, eine mögliche weitere Verschlechterung der GVHD zu verhindern und eine postoperative Rekonstruktion der Immunfunktion bei haploidentischen HSCT-Patienten bereitzustellen.
(3) Invossa-K
Unternehmen: Entwickelt von TissueGene (KolonTissueGene).
Zeit bis zur Markteinführung: Zulassung zur Notierung in Südkorea im Juli 2017.
Indikationen: Zur Behandlung degenerativer Kniearthritis.
Anmerkungen: Invossa-K ist eine allogene Zellgentherapie, an der menschliche Chondrozyten beteiligt sind.Die allogenen Zellen werden in vitro genetisch verändert und die veränderten Zellen können nach intraartikulärer Injektion den transformierenden Wachstumsfaktor β1 (TGF-β1) exprimieren und sezernieren.β1) und verbessert dadurch die Arthrosesymptome.Klinische Ergebnisse zeigen, dass Invossa-K Kniearthrose deutlich verbessern kann.Es wurde 2019 von der koreanischen Lebensmittel- und Arzneimittelbehörde widerrufen, weil der Hersteller die verwendeten Inhaltsstoffe falsch gekennzeichnet hatte.
(4) Zynteglo
Unternehmen: Erforscht und entwickelt von bluebird bio.
Zeit bis zur Markteinführung: Von der Europäischen Union für die Vermarktung im Jahr 2019 und von der FDA für die Vermarktung in den Vereinigten Staaten im August 2022 zugelassen.
Indikationen: Zur Behandlung der transfusionsabhängigen β-Thalassämie.
Anmerkungen: Zynteglo ist eine lentivirale In-vitro-Gentherapie, die eine funktionelle Kopie des normalen β-Globin-Gens (βA-T87Q-Globin-Gen) in hämatopoetische Stammzellen einführt, die dem Patienten über einen lentiviralen Vektor entnommen werden, und diese genetisch veränderten autologen hämatopoetischen Stammzellen dann dem Patienten reinfundiert.Sobald der Patient über ein normales βA-T87Q-Globin-Gen verfügt, kann er normales HbAT87Q-Protein produzieren, wodurch die Notwendigkeit einer Bluttransfusion wirksam reduziert oder ganz beseitigt werden kann.Es handelt sich um eine einmalige Therapie, die lebenslange Bluttransfusionen und lebenslange Medikamente für Patienten ab 12 Jahren ersetzen soll.
(5) Skysona
Unternehmen: Erforscht und entwickelt von bluebird bio.
Zeit bis zur Markteinführung: Im Juli 2021 von der Europäischen Union für die Vermarktung zugelassen und im September 2022 von der FDA für die Vermarktung in den Vereinigten Staaten zugelassen.
Indikationen: Zur Behandlung der frühen zerebralen Adrenoleukodystrophie (CALD).
Anmerkungen: Die Skysona-Gentherapie ist die einzige einmalige Gentherapie, die für die Behandlung der zerebralen Adrenoleukodystrophie (CALD) im Frühstadium zugelassen ist.Skysona (Elivaldogene Autotemcel, Lenti-D) ist eine lentivirale In-vitro-Gentherapie mit hämatopoetischen Stammzellen, Lenti-D.Der allgemeine Ablauf der Therapie ist wie folgt: Dem Patienten werden autologe hämatopoetische Stammzellen entnommen, in vitro durch Lentivirus, das das menschliche ABCD1-Gen trägt, transduziert und modifiziert und dann dem Patienten wieder infundiert.Es wird zur Behandlung von Patienten unter 18 Jahren mit ABCD1-Genmutationen und CALD eingesetzt.
(6) Kymriah
UNTERNEHMEN: Entwickelt von Novartis.
Time to Market: Marktzulassung durch die FDA im August 2017.
Indikationen: Behandlung der akuten lymphoblastischen Vorläufer-B-Zell-Leukämie (ALL) und des rezidivierten und refraktären DLBCL.
Anmerkungen: Kymriah ist ein lentivirales In-vitro-Gentherapie-Medikament, die weltweit erste zur Vermarktung zugelassene CAR-T-Therapie, die auf CD19 abzielt und den co-stimulierenden Faktor 4-1BB nutzt.Der Preis liegt in den USA bei 475.000 US-Dollar und in Japan bei 313.000 US-Dollar.
(7) Yescarta
Unternehmen: Entwickelt von Kite Pharma, einer Tochtergesellschaft von Gilead (GILD).
Zeit bis zur Markteinführung: Marktzulassung durch die FDA im Oktober 2017;Fosun Kite führte die Yescarta-Technologie von Kite Pharma ein und produzierte sie nach Erhalt der Genehmigung in China.Zur Notierung im Land zugelassen.
Indikationen: Zur Behandlung von rezidiviertem oder refraktärem großzelligem B-Zell-Lymphom.
Anmerkungen: Yescarta ist eine retrovirale In-vitro-Gentherapie, die weltweit die zweite zugelassene CAR-T-Therapie ist.Es zielt auf CD19 ab und übernimmt den Costimulator von CD28.Der Preis liegt in den USA bei 373.000 US-Dollar.
(8) Tekartos
Unternehmen: Entwickelt von Gilead (GILD).
Zeit bis zur Markteinführung: Marktzulassung durch die FDA im Juli 2020.
Indikationen: Bei rezidiviertem oder refraktärem Mantelzelllymphom.
Anmerkungen: Tecartus ist eine autologe CAR-T-Zelltherapie, die auf CD19 abzielt, und es ist die dritte CAR-T-Therapie, die weltweit zur Vermarktung zugelassen ist.
(9) Breyanzi
UNTERNEHMEN: Entwickelt von Bristol-Myers Squibb (BMS).
Zeit bis zur Markteinführung: Marktzulassung durch die FDA im Februar 2021.
Indikationen: Rezidiviertes oder refraktäres (R/R) großzelliges B-Zell-Lymphom (LBCL).
Anmerkungen: Breyanzi ist eine In-vitro-Gentherapie auf Basis des Lentivirus, der vierten CAR-T-Therapie, die weltweit zur Vermarktung zugelassen ist und auf CD19 abzielt.Die Zulassung von Breyanzi ist ein Meilenstein für Bristol-Myers Squibb auf dem Gebiet der zellulären Immuntherapie, das das Unternehmen mit der Übernahme von Celgene für 74 Milliarden US-Dollar im Jahr 2019 erworben hat.
(10) Abecma
Unternehmen: Gemeinsam entwickelt von Bristol-Myers Squibb (BMS) und bluebird bio.
Zeit bis zur Markteinführung: Marktzulassung durch die FDA im März 2021.
Indikationen: rezidiviertes oder refraktäres multiples Myelom.
Anmerkungen: Abecma ist eine In-vitro-Gentherapie auf Basis des Lentivirus, die weltweit erste CAR-T-Zelltherapie gegen BCMA und die fünfte von der FDA zugelassene CAR-T-Therapie.Das Prinzip des Medikaments besteht darin, chimäre BCMA-Rezeptoren auf den eigenen T-Zellen des Patienten durch Lentivirus-vermittelte Genmodifikation in vitro zu exprimieren.Behandlung zur Eliminierung nicht genetisch veränderter T-Zellen bei Patienten und anschließende Reinfusion modifizierter T-Zellen, die BCMA-exprimierende Krebszellen bei Patienten suchen und abtöten.
(11) Libmeldy
UNTERNEHMEN: Entwickelt von Orchard Therapeutics.
Zeit bis zur Markteinführung: Zulassung durch die Europäische Union zur Notierung im Dezember 2020.
Indikationen: Zur Behandlung der metachromatischen Leukodystrophie (MLD).
Anmerkungen: Libmeldy ist eine Gentherapie, die auf autologen CD34+-Zellen basiert, die in vitro durch Lentivirus genetisch verändert wurden.Klinische Daten zeigen, dass eine einzelne intravenöse Infusion von Libmeldy den Verlauf einer früh einsetzenden MLD im Vergleich zu schweren motorischen und kognitiven Beeinträchtigungen bei unbehandelten Patienten gleichen Alters wirksam verändern kann.
(12) Benoda
Unternehmen: Entwickelt von WuXi Giant Nuo.
Zeit bis zur Markteinführung: Offiziell von der NMPA im September 2021 zugelassen.
Indikationen: Behandlung erwachsener Patienten mit rezidiviertem oder refraktärem großzelligem B-Zell-Lymphom (r/r LBCL) nach systemischer Zweitlinientherapie oder höher.
Anmerkungen: Benoda ist eine Anti-CD19-CAR-T-Gentherapie und auch das Kernprodukt der WuXi Juro Company.Es ist das zweite in China zugelassene CAR-T-Produkt, mit Ausnahme von rezidiviertem/refraktärem großzelligem B-Zell-Lymphom. WuXi Giant Nuo plant außerdem die Entwicklung einer Regiorensai-Injektion zur Behandlung mehrerer anderer Indikationen, darunter follikuläres Lymphom (FL), Mantelzell-Lymphom (MCL), chronische lymphatische Leukämie (CLL), diffuses großzelliges B-Zell-Lymphom der zweiten Linie (DLBCL) und akute lymphatische Leukämie (ALL).
(13) CARVYKTI
Unternehmen: Das erste zur Vermarktung zugelassene Produkt von Legend Biotech.
Zeit bis zur Markteinführung: Marktzulassung durch die FDA im Februar 2022.
Indikationen: zur Behandlung des rezidivierten oder refraktären multiplen Myeloms (R/R MM).
Anmerkungen: CARVYKTI (Ciltacabtagene Autoleucel, kurz Cilta-cel) ist eine CAR-T-Zell-Immungentherapie mit zwei Einzeldomänen-Antikörpern, die auf das B-Zell-Reifungsantigen (BCMA) abzielen.Daten zeigen, dass CARVYKTI bei Patienten mit rezidiviertem oder refraktärem multiplem Myelom, die zuvor vier oder mehr Therapien erhalten haben (einschließlich Proteasom-Inhibitoren, Immunmodulatoren und monoklonalen Anti-CD38-Antikörpern), eine Gesamtansprechrate von 98 % gezeigt hat.
(14)Ebvallo
UNTERNEHMEN: Entwickelt von Atara Biotherapeutics.
Es handelt sich um die weltweit erste universelle T-Zelltherapie, die im Dezember 2022 von der Europäischen Kommission (EK) zur Vermarktung zugelassen wurde.
Indikationen: Als Monotherapie für die durch das Epstein-Barr-Virus (EBV) bedingte posttransplantationsbedingte lymphoproliferative Erkrankung (EBV+PTLD) muss es sich bei den behandelten Patienten um Erwachsene und Kinder über 2 Jahre handeln, die zuvor mindestens eine andere medikamentöse Therapie erhalten haben.
Anmerkungen: Ebvallo ist eine allogene EBV-spezifische universelle T-Zell-Gentherapie, die auf HLA-beschränkte Weise auf EBV-infizierte Zellen abzielt und diese eliminiert.Die Zulassung dieser Therapie basiert auf den Ergebnissen der entscheidenden klinischen Phase-3-Studie. Die Ergebnisse zeigten, dass die ORR der HCT-Gruppe und der SOT-Gruppe 50 % betrug.Die Rate der vollständigen Remission (CR) betrug 26,3 %, die Rate der teilweisen Remission (PR) betrug 23,7 % und die mittlere Zeit bis zur Remission (TTR) betrug 1,1 Monate.Von den 19 Patienten, die eine Remission erreichten, hatten 11 eine Ansprechdauer (DOR) von mehr als 6 Monaten.Darüber hinaus traten aus Sicherheitsgründen keine Nebenwirkungen wie die Graft-versus-Host-Krankheit (GvHD) oder das Ebvallo-bedingte Zytokinfreisetzungssyndrom auf.
2. In-vivo-Gentherapie auf Basis viraler Vektoren
(1) Gendicine/Jin Sheng
Unternehmen: Entwickelt von der Shenzhen Saibainuo Company.
Zeit bis zur Markteinführung: Zulassung zur Börsennotierung in China im Jahr 2003.
Indikationen: Zur Behandlung von Plattenepithelkarzinomen im Kopf- und Halsbereich.
Hinweis: Die rekombinante humane p53-Adenovirus-Injektion Gendicine/Jinyousheng ist ein Adenovirus-Vektor-Gentherapie-Medikament mit unabhängigen geistigen Eigentumsrechten im Besitz der Shenzhen Saibainuo Company.Das humane Adenovirus Typ 5 besteht aus humanem Adenovirus Typ 5. Ersteres ist die Hauptstruktur für die Antitumorwirkung des Arzneimittels, letzteres fungiert hauptsächlich als Träger.Der Adenovirus-Vektor trägt das therapeutische Gen p53 in die Zielzelle, exprimiert das Tumorsuppressorgen p53 in der Zielzelle und seine Genexpression. Das Produkt kann eine Vielzahl von Antikrebsgenen hochregulieren und die Aktivitäten einer Vielzahl von Onkogenen herunterregulieren, wodurch die Tumorsuppressorwirkung des Körpers verstärkt und der Zweck der Tumortötung erreicht wird.
(2) Rigvir
Unternehmen: Entwickelt von Latima Company, Lettland.
Zeitpunkt der Listung: 2004 zur Listung in Lettland zugelassen.
Indikationen: Zur Behandlung von Melanomen.
Anmerkungen: Rigvir ist eine Gentherapie, die auf einem genetisch veränderten ECHO-7-Enterovirus-Vektor basiert.Derzeit ist das Medikament in Lettland, Estland, Polen, Armenien, Weißrussland usw. eingeführt und wird auch in EU-Ländern bei der EMA registriert.Klinische Fälle in den letzten zehn Jahren haben gezeigt, dass das onkolytische Rigvir-Virus sicher und wirksam ist und die Überlebensrate von Melanompatienten um das 4- bis 6-fache erhöhen kann.Darüber hinaus ist diese Therapie auch bei einer Vielzahl anderer Krebsarten anwendbar, darunter Darmkrebs, Bauchspeicheldrüsenkrebs, Blasenkrebs, Nierenkrebs, Prostatakrebs, Lungenkrebs, Gebärmutterkrebs, Lymphosarkom usw.
(3) Oncorin
Unternehmen: Entwickelt von der Shanghai Sanwei Biological Company.
Zeit bis zur Markteinführung: Zulassung zur Börsennotierung in China im Jahr 2005.
Indikationen: Behandlung von Kopf-Hals-Tumoren, Leberkrebs, Bauchspeicheldrüsenkrebs, Gebärmutterhalskrebs und anderen Krebsarten.
Anmerkungen: Oncorine (安科瑞) ist ein Gentherapieprodukt für onkolytische Viren, das Adenoviren als Träger verwendet.Es wird ein onkolytisches Adenovirus gewonnen, das sich spezifisch in p53-Gen-defizienten oder abnormalen Tumoren replizieren kann, was zur Lyse von Tumorzellen führt und dadurch Tumorzellen abtötet.ohne normale Zellen zu schädigen.Klinische Studien haben gezeigt, dass Ankerui eine gute Sicherheit und Wirksamkeit bei einer Vielzahl bösartiger Tumoren aufweist.
(4) Glybera
Unternehmen: Entwickelt von uniQure.
Time to Market: Zulassung zur Notierung in Europa im Jahr 2012.
Indikationen: Behandlung des Lipoprotein-Lipase-Mangels (LPLD) mit schweren oder wiederkehrenden Episoden einer Pankreatitis trotz streng fettreduzierter Ernährung.
Anmerkungen: Glybera (Alipogen Tiparvovec) ist ein auf AAV basierendes Gentherapie-Medikament, das AAV als Träger verwendet, um das therapeutische Gen LPL in Muskelzellen zu transduzieren, sodass die entsprechenden Zellen eine bestimmte Menge an Lipoproteinlipase produzieren können. Um die Krankheit zu lindern, ist diese Therapie nach der Verabreichung über einen langen Zeitraum wirksam (Arzneimittelwirkung kann viele Jahre anhalten).Das Medikament wurde 2017 vom Markt genommen. Der Grund für die Rücknahme kann auf zwei Faktoren zurückzuführen sein: hohe Preise und begrenzte Marktnachfrage.Die durchschnittlichen Behandlungskosten des Medikaments belaufen sich auf bis zu 1 Million US-Dollar, und bisher hat nur ein Patient es gekauft und verwendet.Obwohl die Krankenversicherung dafür 900.000 US-Dollar erstattet hat, stellt dies auch eine relativ große Belastung für die Versicherungsgesellschaft dar.Darüber hinaus sind die Indikationen, auf die das Medikament abzielt, mit einer Inzidenzrate von etwa 1 zu 1 Million und einer hohen Rate an Fehldiagnosen zu selten.
(5) Imlygisch
Unternehmen: Entwickelt von Amgen.
Zeit bis zur Markteinführung: Im Jahr 2015 wurde die Zulassung in den Vereinigten Staaten und der Europäischen Union erteilt.
Indikationen: Behandlung von Melanomläsionen, die durch eine Operation nicht vollständig entfernt werden können.
Anmerkungen: Imlygic ist ein abgeschwächtes Herpes-simplex-Virus Typ 1, das durch Gentechnik verändert wurde (durch Löschung seiner ICP34.5- und ICP47-Genfragmente und Einfügen des menschlichen Granulozyten-Makrophagen-Kolonie-stimulierenden Faktors GM-CSF-Gens in das Virus). Das onkolytische Virus (HSV-1) ist die erste von der FDA zugelassene onkolytische Virus-Gentherapie.Die Verabreichungsmethode ist eine intraläsionale Injektion, die direkt in Melanomläsionen injiziert werden kann, um das Aufbrechen von Tumorzellen zu bewirken, von Tumoren abgeleitete Antigene und GM-CSF freizusetzen und Antitumor-Immunreaktionen zu fördern.
(6) Luxturna
Unternehmen: Entwickelt von Spark Therapeutics, einer Tochtergesellschaft von Roche.
Zeit bis zur Markteinführung: Es wurde 2017 von der FDA für die Vermarktung zugelassen und 2018 dann für die Vermarktung in Europa zugelassen.
Indikationen: Zur Behandlung von Kindern und Erwachsenen, die aufgrund von Doppelkopie-RPE65-Genmutationen ihr Sehvermögen verloren haben, aber über eine ausreichende Anzahl lebensfähiger Netzhautzellen verfügen.
Anmerkungen: Luxturna ist eine AAV-basierte Gentherapie, die durch subretinale Injektion verabreicht wird.Die Gentherapie verwendet AAV2 als Träger, um eine funktionelle Kopie des normalen RPE65-Gens in die Netzhautzellen des Patienten einzuführen, sodass die entsprechenden Zellen das normale RPE65-Protein exprimieren, wodurch der RPE65-Proteinmangel des Patienten ausgeglichen und dadurch das Sehvermögen des Patienten verbessert wird.
(7) Zolgensma
Unternehmen: Entwickelt von AveXis, einer Tochtergesellschaft von Novartis.
Time to Market: Marktzulassung durch die FDA im Mai 2019.
Indikationen: Behandlung von Patienten mit spinaler Muskelatrophie (SMA) unter 2 Jahren.
Anmerkungen: Zolgensma ist eine Gentherapie, die auf dem AAV-Vektor basiert.Dieses Medikament ist der einzige einmalige Behandlungsplan für spinale Muskelatrophie, der weltweit zur Vermarktung zugelassen ist.Die Einführung des Medikaments eröffnet eine neue Ära in der Behandlung der spinalen Muskelatrophie.Seite, ist ein Meilenstein Fortschritt.Diese Gentherapie verwendet den scAAV9-Vektor, um dem Patienten das normale SMN1-Gen durch intravenöse Infusion einzuführen, um normales SMN1-Protein zu produzieren und dadurch die Funktion betroffener Zellen wie Motoneuronen zu verbessern.Im Gegensatz dazu erfordern die SMA-Medikamente Spinraza und Evrysdi eine wiederholte Langzeitdosierung.Spinraza wird alle vier Monate als Injektion in die Wirbelsäule verabreicht, und Evrysdi ist ein oral einzunehmendes Medikament.
(8) Delytact
Unternehmen: Entwickelt von Daiichi Sankyo Company Limited (TYO: 4568).
Zeit bis zur Markteinführung: Bedingte Genehmigung durch das japanische Ministerium für Gesundheit, Arbeit und Soziales (MHLW) im Juni 2021.
Indikationen: Zur Behandlung von malignen Gliomen.
Anmerkungen: Delytact ist das vierte weltweit zugelassene Gentherapieprodukt für onkolytische Viren und das erste für die Behandlung von malignen Gliomen zugelassene Produkt für onkolytische Viren.Delytact ist ein gentechnisch verändertes onkolytisches Virus des Herpes-simplex-Virus Typ 1 (HSV-1), das von Dr. Todo und Kollegen entwickelt wurde.Delytact führt zusätzliche Deletionsmutationen in das G207-Genom des HSV-1 der zweiten Generation ein, wodurch dessen selektive Replikation in Krebszellen und die Induktion von Antitumor-Immunantworten bei gleichzeitig hoher Sicherheit verbessert werden.Delytact ist das erste onkolytische HSV-1 der dritten Generation, das sich derzeit in der klinischen Prüfung befindet.Die Zulassung von Delytact in Japan basiert hauptsächlich auf einer einarmigen klinischen Phase-2-Studie.Bei Patienten mit rezidivierendem Glioblastom erreichte Delytact den primären Endpunkt der Ein-Jahres-Überlebensrate, und die Ergebnisse zeigten, dass Delytact im Vergleich zu G207 eine bessere Wirksamkeit zeigte.Starke Replikationskraft und höhere Antitumoraktivität.Dies war in soliden Tumormodellen von Brust-, Prostata-, Schwannom-, Nasopharynx-, hepatozellulärem, kolorektalem, bösartigem Tumor der peripheren Nervenscheide und Schilddrüsenkrebs wirksam.
(9) Upstaza
UNTERNEHMEN: Entwickelt von PTC Therapeutics, Inc. (NASDAQ: PTCT).
Zeit bis zur Markteinführung: Zulassung durch die Europäische Union zur Vermarktung im Juli 2022.
Indikationen: Bei Mangel an aromatischer L-Aminosäure-Decarboxylase (AADC) ist es für die Behandlung von Patienten ab 18 Monaten zugelassen.
Anmerkungen: Upstaza™ (Eladocagene Exuparvovec) ist eine In-vivo-Gentherapie mit dem Adeno-assoziierten Virus Typ 2 (AAV2) als Träger.Patienten werden aufgrund von Mutationen im Gen, das für das AADC-Enzym kodiert, krank.AAV2 trägt ein gesundes Gen, das für das AADC-Enzym kodiert.Durch die Form der Genkompensation wird eine therapeutische Wirkung erzielt.Theoretisch ist eine Verabreichung über einen langen Zeitraum wirksam.Es ist die erste vermarktete Gentherapie, die direkt in das Gehirn injiziert wird.Die Marktzulassung gilt für alle 27 EU-Mitgliedstaaten sowie Island, Norwegen und Liechtenstein.
(10) Roctavian
Unternehmen: Entwickelt von BioMarin Pharmaceutical (BioMarin).
Zeit bis zur Markteinführung: Marktzulassung durch die Europäische Union im August 2022;Marktzulassung durch die UK Medicines and Healthcare Products Administration (MHRA) im November 2022.
Indikationen: Zur Behandlung erwachsener Patienten mit schwerer Hämophilie A, bei denen in der Vorgeschichte keine Hemmung des FVIII-Faktors aufgetreten ist und die keine AAV5-Antikörper aufweisen.
Anmerkungen: Roctavian (Valoctocogene roxaparvovec) verwendet AAV5 als Vektor und den menschlichen leberspezifischen Promotor HLP, um die Expression des menschlichen Gerinnungsfaktors VIII (FVIII) mit deletierter B-Domäne voranzutreiben.Die Entscheidung der Europäischen Kommission, die Vermarktung von Valoctocogen Roxaparvovec zu genehmigen, basiert auf den Gesamtdaten des klinischen Entwicklungsprojekts des Arzneimittels.Unter anderem zeigten die Ergebnisse der klinischen Phase-III-Studie GENER8-1, dass im Vergleich zu den Daten des Jahres vor der Einschreibung nach einer einzelnen Infusion von Valoctocogen Roxaparvovec die jährliche Blutungsrate (ABR) des Probanden deutlich reduziert, die Häufigkeit der Verwendung von rekombinanten Gerinnungsfaktor VIII (F8)-Proteinpräparaten reduziert oder die Aktivität von F8 im Blut im Körper deutlich erhöht ist.Nach 4-wöchiger Behandlung waren die jährliche F8-Verwendungsrate und die behandlungsbedürftige ABR des Probanden um 99 % bzw. 84 % reduziert, und der Unterschied war statistisch signifikant (p < 0,001).Das Sicherheitsprofil war gut und bei keinem Probanden traten F8-Faktor-Hemmung, maligne oder thrombosebedingte Nebenwirkungen auf, und es wurden keine behandlungsbedingten schwerwiegenden unerwünschten Ereignisse (SAEs) gemeldet.
(11) Hemgenix
Unternehmen: Entwickelt von UniQure Corporation.
Zeit bis zur Markteinführung: Zulassung durch die FDA zur Vermarktung im November 2022.
Indikationen: Zur Behandlung erwachsener Patienten mit Hämophilie B.
Anmerkungen: Hemgenix ist eine Gentherapie, die auf dem AAV5-Vektor basiert.Das Medikament ist mit der Genvariante FIX-Padua des Gerinnungsfaktors IX (FIX) ausgestattet, die intravenös verabreicht wird.Nach der Verabreichung kann das Gen den FIX-Gerinnungsfaktor in der Leber exprimieren und absondern. Nachdem es in das Blut gelangt ist, um die Gerinnungsfunktion auszuüben, um den Zweck der Behandlung zu erreichen, ist theoretisch eine Verabreichung über einen langen Zeitraum wirksam.
(12) Adstiladrin
Unternehmen: Entwickelt von Ferring Pharmaceuticals.
Zeit bis zur Markteinführung: Zulassung durch die FDA zur Vermarktung im Dezember 2022.
Indikationen: Zur Behandlung von hochriskantem nicht-muskelinvasivem Blasenkrebs (NMIBC), der nicht auf Bacillus Calmette-Guerin (BCG) anspricht.
Anmerkungen: Adstiladrin ist eine Gentherapie, die auf einem nicht replizierenden adenoviralen Vektor basiert, der das Interferon alfa-2b-Protein in Zielzellen überexprimieren kann und über einen Harnkatheter in die Blase verabreicht wird (einmal alle drei Monate verabreicht). Der Virusvektor kann effektiv die Zellen der Blasenwand infizieren und dann das Interferon alfa-2b-Protein überexprimieren, um eine therapeutische Wirkung auszuüben.Diese neuartige Gentherapiemethode verwandelt somit die eigenen Blasenwandzellen des Patienten in eine Miniatur-„Fabrik“, die Interferon produziert, und verbessert so die Fähigkeit des Patienten, Krebs zu bekämpfen.
Die Sicherheit und Wirksamkeit von Adstiladrin wurden in einer multizentrischen klinischen Studie mit 157 Patienten mit Hochrisiko-BCG-nicht-responsivem NMIBC untersucht.Die Patienten erhielten Adstiladrin alle drei Monate über einen Zeitraum von bis zu 12 Monaten oder bis eine inakzeptable toxische Wirkung auf die Behandlung oder ein erneutes Auftreten von hochgradigem NMIBC auftrat.Insgesamt erreichten 51 Prozent der mit Adstiladrin behandelten Patienten ein vollständiges Ansprechen (Verschwinden aller bei Zystoskopie, Biopsiegewebe und Urin sichtbaren Anzeichen von Krebs).
3. Kleine Nukleinsäure-Medikamente
(1) Vitraven
Unternehmen: Gemeinsam entwickelt von Ionis Pharma (ehemals Isis Pharma) und Novartis.
Zeit bis zur Markteinführung: 1998 und 1999 wurde es von der FDA und der EU-EMA für die Vermarktung zugelassen.
Indikationen: Zur Behandlung der Cytomegalovirus-Retinitis bei HIV-positiven Patienten.
Anmerkungen: Vitravene ist ein Antisense-Oligonukleotid-Medikament und das erste Oligonukleotid-Medikament, das weltweit zur Vermarktung zugelassen wurde.In der Anfangsphase der Börsennotierung war die Marktnachfrage nach Anti-CMV-Medikamenten sehr dringend;Später ging die Zahl der CMV-Fälle aufgrund der Entwicklung einer hochaktiven antiretroviralen Therapie stark zurück.Aufgrund der schleppenden Marktnachfrage wurde das Medikament 2002 auf den Markt gebracht und 2006 in den EU-Ländern und den USA zurückgezogen.
(2) Macugen
Unternehmen: Gemeinsam entwickelt von Pfizer und Eyetech.
Zeit bis zur Markteinführung: Zulassung zur Notierung in den Vereinigten Staaten im Jahr 2004.
Indikationen: Zur Behandlung der neovaskulären altersbedingten Makuladegeneration.
Anmerkungen: Macugen ist ein pegyliertes modifiziertes Oligonukleotid-Medikament, das auf den vaskulären endothelialen Wachstumsfaktor (VEGF165-Subtyp) abzielen und ihn binden kann. Die Verabreichungsmethode ist die intravitreale Injektion.
(3) Defitelio
Unternehmen: Entwickelt von Jazz Pharmaceuticals.
Zeit bis zur Markteinführung: Es wurde 2013 von der Europäischen Union zur Vermarktung zugelassen und im März 2016 von der FDA zur Vermarktung zugelassen.
Indikationen: Zur Behandlung von Lebervenenverschlusskrankheiten im Zusammenhang mit Nieren- oder Lungenfunktionsstörungen nach einer hämatopoetischen Stammzelltransplantation.
Anmerkungen: Defitelio ist ein Oligonukleotid-Medikament, bei dem es sich um eine Mischung aus Oligonukleotiden mit Plasmineigenschaften handelt.2009 aus kommerziellen Gründen vom Markt genommen.
(4) Kynamro
Unternehmen: Gemeinsam entwickelt von Ionis Pharma und Kastle.
Zeit bis zur Markteinführung: Im Jahr 2013 wurde es in den USA als Orphan Drug für die Vermarktung zugelassen.
Indikationen: Zur adjuvanten Behandlung der homozygoten familiären Hypercholesterinämie.
Anmerkungen: Kynamro ist ein Antisense-Oligonukleotid-Medikament, bei dem es sich um ein Antisense-Oligonukleotid handelt, das auf menschliche Apo B-100-mRNA abzielt.Kynamro wird einmal wöchentlich in einer Dosierung von 200 mg subkutan verabreicht.
(5) Spinraza
Unternehmen: Entwickelt von Ionis Pharmaceuticals.
Time to Market: Marktzulassung durch die FDA im Dezember 2016.
Indikationen: Zur Behandlung der spinalen Muskelatrophie (SMA).
Anmerkungen: Spinraza (Nusinersen) ist ein Antisense-Oligonukleotid-Medikament.Durch die Bindung an die Schnittstelle von SMN2-Exon 7 kann Spinraza die RNA-Spaltung des SMN2-Gens verändern und so die Produktion von voll funktionsfähigem SMN-Protein steigern.Im August 2016 übte BIOGEN seine Option aus, die weltweiten Rechte an Spinraza zu erwerben.Spinraza begann seine erste klinische Studie am Menschen erst im Jahr 2011. In nur fünf Jahren wurde es 2016 von der FDA für die Vermarktung zugelassen, was die volle Anerkennung seiner Wirksamkeit durch die FDA widerspiegelt.Das Medikament wurde im April 2019 für die Vermarktung in China zugelassen. Der gesamte Zulassungszyklus für Spinraza in China dauerte weniger als sechs Monate, und seit der ersten Zulassung von Spinraza in den USA sind zwei Jahre und zwei Monate vergangen.Die Geschwindigkeit der Börsennotierung in China ist bereits sehr hoch.Dies ist auch auf die Tatsache zurückzuführen, dass das Center for Drug Evaluation am 1. November 2018 die „Mitteilung zur Veröffentlichung der Liste der ersten Charge von in der klinischen Praxis dringend benötigten neuen Arzneimitteln aus Übersee“ herausgab und in die erste Charge von 40 ausländischen neuen Arzneimitteln zur beschleunigten Prüfung aufgenommen wurde, zu denen auch Spinraza zählte.
(6) Exondys 51
Unternehmen: Entwickelt von AVI BioPharma (später umbenannt in Sarepta Therapeutics).
Zeit bis zur Markteinführung: Im September 2016 wurde es von der FDA zur Vermarktung zugelassen.
Indikationen: Zur Behandlung der Duchenne-Muskeldystrophie (DMD) mit Exon-51-Skipping-Gen-Mutation im DMD-Gen.
Anmerkungen: Exondys 51 ist ein Antisense-Oligonukleotid-Medikament. Das Antisense-Oligonukleotid kann an die Position von Exon 51 der Prä-mRNA des DMD-Gens binden, was zur Bildung von reifer mRNA führt, ein Teil von Exon 51 wird blockiert. Durch die Exzision wird der mRNA-Leserahmen teilweise korrigiert und Patienten können einige funktionelle Formen von Dystrophin synthetisiert werden, die kürzer als das normale Protein sind, wodurch sich die Symptome des Patienten verbessern.
(7) Tegsedi
Unternehmen: Entwickelt von Ionis Pharmaceuticals.
Zeit bis zur Markteinführung: Im Juli 2018 wurde es von der Europäischen Union zur Vermarktung zugelassen.
Indikationen: Zur Behandlung der hereditären Transthyretin-Amyloidose (hATTR).
Anmerkungen: Tegsedi ist ein Antisense-Oligonukleotid-Medikament, das auf Transthyretin-mRNA abzielt.Es ist das erste Medikament weltweit, das zur Behandlung von hATTR zugelassen ist.Es wird durch subkutane Injektion verabreicht.Das Medikament reduziert die Produktion des ATTR-Proteins, indem es auf die mRNA von Transthyretin (ATTR) abzielt, und weist ein gutes Nutzen-Risiko-Verhältnis bei der Behandlung von ATTR auf. Die Neuropathie und Lebensqualität des Patienten wurden erheblich verbessert und es ist mit TTR-Mutationstypen kompatibel. Weder das Krankheitsstadium noch das Vorliegen einer Kardiomyopathie waren relevant.
(8) Onpattro
Unternehmen: Gemeinsam entwickelt von Alnylam Corporation und Sanofi Corporation.
Zeit bis zur Markteinführung: Zulassung zur Notierung in den Vereinigten Staaten im Jahr 2018.
Indikationen: Zur Behandlung der hereditären Transthyretin-Amyloidose (hATTR).
Anmerkungen: Onpattro ist ein siRNA-Medikament, das auf die Transthyretin-mRNA abzielt. Es reduziert die Produktion des ATTR-Proteins in der Leber und reduziert die Ansammlung von Amyloidablagerungen in peripheren Nerven, indem es auf die mRNA von Transthyretin (ATTR) abzielt, wodurch die Krankheitssymptome verbessert und gelindert werden.
(9) Givlaari
Unternehmen: Entwickelt von Alnylam Corporation.
Time to Market: Marktzulassung durch die FDA im November 2019.
Indikationen: Zur Behandlung der akuten hepatischen Porphyrie (AHP) bei Erwachsenen.
Anmerkungen: Givlaari ist ein siRNA-Medikament und nach Onpattro das zweite zur Vermarktung zugelassene siRNA-Medikament.Die Art der Verabreichung ist eine subkutane Injektion.Das Medikament zielt auf die mRNA des ALAS1-Proteins ab, und eine monatliche Behandlung mit Givlaari kann den ALAS1-Spiegel in der Leber deutlich und dauerhaft senken, wodurch die Spiegel von neurotoxischem ALA und PBG auf den normalen Bereich gesenkt werden und dadurch die Krankheitssymptome des Patienten gelindert werden.Die Daten zeigten, dass bei Patienten, die mit Givlaari behandelt wurden, die Anzahl der Anfälle im Vergleich zur Placebogruppe um 74 % zurückging.
(10) Vyondys53
UNTERNEHMEN: Entwickelt von Sarepta Therapeutics.
Time to Market: Marktzulassung durch die FDA im Dezember 2019.
Indikationen: Zur Behandlung von DMD-Patienten mit Dystrophin-Gen-Exon-53-Splicing-Mutation.
Anmerkungen: Vyondys 53 ist ein Antisense-Oligonukleotid-Medikament, das auf den Spleißprozess der Dystrophin-Prä-mRNA abzielt.Exon 53 ist teilweise verkürzt, also nicht auf der reifen mRNA vorhanden, und soll ein verkürztes, aber immer noch funktionsfähiges Dystrophin produzieren und dadurch die körperliche Leistungsfähigkeit der Patienten verbessern.
(11) Waylivra
Unternehmen: Entwickelt von Ionis Pharmaceuticals und seiner Tochtergesellschaft Akcea Therapeutics.
Time to Market: Die Marktzulassung erfolgte im Mai 2019 durch die Europäische Arzneimittel-Agentur (EMA).
Indikationen: Als adjuvante Therapie zusätzlich zur Diätkontrolle bei erwachsenen Patienten mit familiärem Chylomikronämie-Syndrom (FCS).
Anmerkungen: Waylivra ist ein Antisense-Oligonukleotid-Medikament und weltweit das erste zugelassene Medikament zur Behandlung von FCS.
(12) Leqvio
Unternehmen: Entwickelt von Novartis.
Zeit bis zur Markteinführung: Zulassung durch die Europäische Union zur Vermarktung im Dezember 2020.
Indikationen: Zur Behandlung von Erwachsenen mit primärer Hypercholesterinämie (heterozygot familiär und nicht familiär) oder gemischter Dyslipidämie.
Anmerkungen: Leqvio ist ein siRNA-Medikament, das auf PCSK9-mRNA abzielt.Es handelt sich um die weltweit erste siRNA-Therapie zur Senkung des Cholesterinspiegels (LDL-C).Es wird durch subkutane Injektion verabreicht.Das Medikament reduziert den Spiegel des PCSK9-Proteins durch RNA-Interferenz und senkt dadurch den LDL-C-Spiegel.Klinische Daten zeigen, dass Leqvio bei Patienten, die den LDL-C-Spiegel nach der Behandlung mit der maximal verträglichen Statindosis nicht auf den Zielwert senken können, den LDL-C-Spiegel um etwa 50 % senken kann.
(13)Oxlumo
Unternehmen: Entwickelt von Alnylam Pharmaceuticals.
Zeit bis zur Markteinführung: Zulassung durch die Europäische Union zur Vermarktung im November 2020.
Indikationen: Zur Behandlung der primären Hyperoxalurie Typ 1 (PH1).
Anmerkungen: Oxlumo ist ein siRNA-Medikament, das auf die mRNA der Hydroxysäureoxidase 1 (HAO1) abzielt, und die Verabreichungsmethode ist eine subkutane Injektion.Das Medikament wurde unter Verwendung der neuesten verbesserten Stabilisierungschemie von Alnylam, der ESC-GalNAc-Konjugationstechnologie, entwickelt, die eine subkutan verabreichte siRNA mit größerer Persistenz und Wirksamkeit ermöglicht.Das Medikament baut die Hydroxysäureoxidase 1 (HAO1)-mRNA ab oder hemmt sie, senkt den Glykolatoxidasespiegel in der Leber und verbraucht dann das für die Oxalatproduktion erforderliche Substrat, wodurch die Oxalatproduktion reduziert wird, um das Fortschreiten der Krankheit bei Patienten zu kontrollieren und die Krankheitssymptome zu verbessern.
(14) Viltepso
Unternehmen: Entwickelt von NS Pharma, einer Tochtergesellschaft von Nippon Shinyaku.
Zeit bis zur Markteinführung: Marktzulassung durch die FDA im August 2020.
Indikationen: Zur Behandlung der Duchenne-Muskeldystrophie (DMD) mit Exon-53-Skipping-Gen-Mutation im DMD-Gen.
Anmerkungen: Viltepso ist ein Antisense-Oligonukleotid-Medikament, das an die Position von Exon 53 der Prä-mRNA des DMD-Gens binden kann, wodurch ein Teil von Exon 53 nach der Bildung reifer mRNA herausgeschnitten wird, wodurch der mRNA-Leserahmen teilweise korrigiert wird. Die Box hilft Patienten, einige funktionelle Formen von Dystrophin zu synthetisieren, die kürzer als normale Proteine sind, wodurch die Symptome der Patienten verbessert werden.
(15) Amondys 45
Unternehmen: Entwickelt von Sarepta Therapeutics.
Zeit bis zur Markteinführung: Marktzulassung durch die FDA im Februar 2021.
Indikationen: Zur Behandlung der Duchenne-Muskeldystrophie (DMD) mit Exon-45-Skipping-Gen-Mutation im DMD-Gen.
Anmerkungen: Amondys 45 ist ein Antisense-Oligonukleotid-Medikament. Das Antisense-Oligonukleotid kann an die Position von Exon 45 der Prä-mRNA des DMD-Gens binden, was dazu führt, dass der Teil von Exon 45 nach der Bildung reifer mRNA blockiert wird. Exzision, wodurch der mRNA-Leserahmen teilweise korrigiert wird, was Patienten hilft, einige funktionelle Formen von Dystrophin zu synthetisieren, das kürzer als das normale Protein ist, wodurch die Symptome des Patienten verbessert werden.
(16) Amvuttra (Vutrisiran)
Unternehmen: Entwickelt von Alnylam Pharmaceuticals.
Zeit bis zur Markteinführung: Marktzulassung durch die FDA im Juni 2022.
Indikationen: Zur Behandlung der hereditären Transthyretin-Amyloidose mit Polyneuropathie (hATTR-PN) bei Erwachsenen.
Anmerkungen: Amvuttra (Vutrisiran) ist ein siRNA-Medikament, das auf Transthyretin (ATTR)-mRNA abzielt und durch subkutane Injektion verabreicht wird.Vutrisiran basiert auf dem Design der Enhanced Stability Chemistry (ESC)-GalNAc-Konjugat-Abgabeplattform von Alnylam mit erhöhter Wirksamkeit und metabolischer Stabilität.Die Zulassung der Therapie basiert auf den 9-Monats-Daten der klinischen Phase-III-Studie (HELIOS-A), und die Gesamtergebnisse zeigen, dass die Therapie die Symptome von hATTR-PN verbesserte und der Zustand der Patienten bei mehr als 50 % umgekehrt oder nicht mehr verschlechtert wurde.
4. Andere Gentherapeutika
(1) Rexin-G
Unternehmen: Entwickelt von Epeius Biotech.
Zeit bis zur Markteinführung: Im Jahr 2005 wurde es von der philippinischen Lebensmittel- und Arzneimittelbehörde (BFAD) zur Vermarktung zugelassen.
Indikationen: Zur Behandlung fortgeschrittener Krebserkrankungen, die gegen eine Chemotherapie resistent sind.
Anmerkungen: Rexin-G ist eine genbeladene Nanopartikel-Injektion.Es führt das mutierte Gen Cyclin G1 über einen retroviralen Vektor in die Zielzellen ein, um solide Tumoren gezielt abzutöten.Die Verabreichungsmethode ist die intravenöse Infusion.Da es sich um ein auf den Tumor gerichtetes Medikament handelt, das aktiv nach metastasierenden Krebszellen sucht und diese zerstört, hat es eine gewisse heilende Wirkung auf Patienten, bei denen andere Krebsmedikamente, einschließlich zielgerichteter Biologika, versagt haben.
(2) Neovasculgen
Unternehmen: Entwickelt vom Institut für menschliche Stammzellen.
Zeitpunkt der Notierung: Die Notierung in Russland wurde am 7. Dezember 2011 genehmigt und 2013 in der Ukraine eingeführt.
Indikationen: Zur Behandlung peripherer arterieller Gefäßerkrankungen, einschließlich schwerer Extremitätenischämie.
Anmerkungen: Neovasculgen ist eine Gentherapie auf Basis von DNA-Plasmiden.Das Gen für den vaskulären endothelialen Wachstumsfaktor (VEGF) 165 wird auf dem Plasmidrückgrat konstruiert und den Patienten infundiert.
(3) Collategene
Unternehmen: Gemeinsam entwickelt von der Universität Osaka und Risikokapitalgesellschaften.
Zeit bis zur Markteinführung: Genehmigt durch das japanische Ministerium für Gesundheit, Arbeit und Soziales im August 2019.
Indikationen: Behandlung kritischer Ischämie der unteren Extremitäten.
Anmerkungen: Collategene ist eine plasmidbasierte Gentherapie, das erste inländische Gentherapie-Medikament, das von AnGes, einem Gentherapie-Unternehmen in Japan, hergestellt wird.Der Hauptbestandteil dieses Arzneimittels ist ein nacktes Plasmid, das die Gensequenz des menschlichen Hepatozytenwachstumsfaktors (HGF) enthält.Wenn das Medikament in die Muskeln der unteren Gliedmaßen injiziert wird, fördert der exprimierte HGF die Bildung neuer Blutgefäße um die verschlossenen Blutgefäße herum.Klinische Studien haben seine Wirkung auf die Verbesserung von Geschwüren bestätigt.
Wie kann Foregene die Entwicklung der Gentherapie unterstützen?
Wir tragen dazu bei, Screening-Zeit bei groß angelegten Screenings in der frühen Phase der siRNA-Arzneimittelentwicklung zu sparen.
Weitere Details finden Sie unter:
https://www.foreivd.com/cell-direct-rt-qpcr-kit-direct-rt-qpcr-series/
Zeitpunkt der Veröffentlichung: 27. Dezember 2022



