



Hörverlust (HL) ist die häufigste Sinnesbehinderung beim Menschen.In entwickelten Ländern werden etwa 80 % der Fälle von prälingualer Taubheit bei Kindern durch genetische Faktoren verursacht.Am häufigsten sind einzelne Gendefekte (wie in Abb. 1 dargestellt), 124 Genmutationen sind mit nicht-syndrolem Hörverlust beim Menschen verbunden, der Rest wird durch Umweltfaktoren verursacht.Ein Cochlea-Implantat (ein im Innenohr platziertes elektronisches Gerät, das den Hörnerv direkt elektrisch stimuliert) ist bei weitem die wirksamste Option zur Behandlung von schwerem HL, während ein Hörgerät (ein externes elektronisches Gerät, das Schallwellen umwandelt und verstärkt) Patienten mit mittelschwerem HL helfen kann.Allerdings stehen derzeit keine Medikamente zur Behandlung des erblichen HL (GHL) zur Verfügung.In den letzten Jahren hat die Gentherapie als vielversprechender Ansatz zur Behandlung von Innenohrstörungen zunehmend Beachtung gefunden.
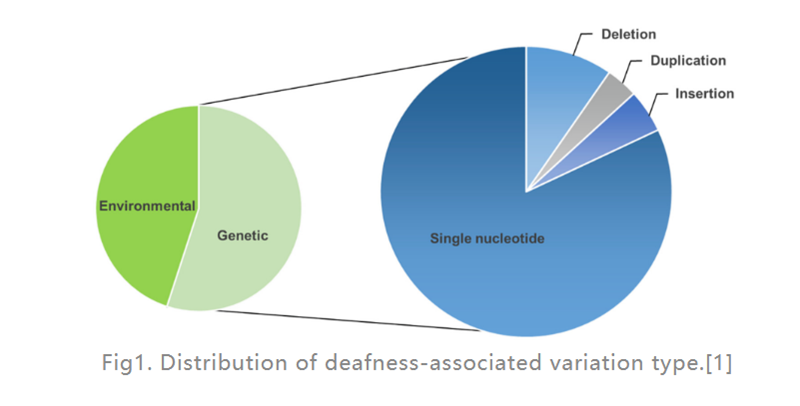
Abb. 1.Verteilung des mit Taubheit assoziierten Variationstyps.[1]
Kürzlich veröffentlichten Wissenschaftler des Salk Institute und der University of Sheffield in Molecular Therapy – Methods & Clinical Development [2] ein Forschungsergebnis, das breite Anwendungsaussichten für die In-vivo-Gentherapie erblicher Taubheit zeigte.Uri Manor, Assistenzprofessor für Forschung am Salk Institute und Direktor des Waitt Center for Advanced Biophotonics, sagte, er sei mit schwerem Hörverlust geboren worden und habe das Gefühl, dass die Wiederherstellung des Hörvermögens ein wunderbares Geschenk sei.Seine früheren Forschungen ergaben, dass Eps8 ein Aktin-regulierendes Protein mit Aktin-Bindungs- und Capping-Aktivitäten ist;In Cochlea-Haarzellen kommt der von Eps8 mit MYO15A, WHIRLIN, GPSM2 und GNAI3 gebildete Proteinkomplex hauptsächlich in den meisten Haarbündeln vor. Die Spitzen langer Stereozilien, die zusammen mit MYO15A BAIAP2L2 an den Spitzen kürzerer Stereozilien lokalisieren, sind für die Aufrechterhaltung von Haarbündeln erforderlich.Daher kann Eps8 die Länge der Stereozilien der Haarzellen regulieren, was für eine normale Hörfunktion unerlässlich ist;Die Deletion oder Mutation von Eps8 führt zu kurzen Stereozilien, die es unmöglich machen, Schall in elektrische Signale für die Gehirnwahrnehmung umzuwandeln, was wiederum zu Taubheit führt..Gleichzeitig stellte der Mitarbeiter Walter Marcotti, Professor an der Universität Sheffield, fest, dass sich Haarzellen ohne Eps8 nicht normal entwickeln können.In dieser Studie untersuchten Manor und Marcotti gemeinsam, ob die Zugabe von Eps8 zu Stereoziliarzellen deren Funktion wiederherstellen und dadurch das Hörvermögen bei Mäusen verbessern könnte.Das Forschungsteam verwendete den Adeno-assoziierten Virus (AAV)-Vektor Anc80L65, um die kodierende Sequenz, die Wildtyp-EPS8 enthält, durch Rundfenstermembraninjektion in die Cochlea von Eps8-/- neugeborenen P1-P2-Mäusen zu transportieren;in Cochlea-Haarzellen von Mäusen Die Funktion der Stereozilien wurde vor ihrer Reifung wiederhergestellt;und der Reparatureffekt wurde durch bildgebende Technologie und Messung von Stereozilien charakterisiert.Die Ergebnisse zeigten, dass Eps8 die Länge der Stereozilien erhöhte und die Funktion der Haarzellen in Niederfrequenzzellen wiederherstellte.Sie fanden auch heraus, dass die Zellen mit der Zeit scheinbar ihre Fähigkeit verloren, durch diese Gentherapie gerettet zu werden.Die Schlussfolgerung ist, dass diese Behandlung möglicherweise in der Gebärmutter verabreicht werden muss, da die Eps8-/-Haarzellen nach der Geburt der Mäuse möglicherweise gereift sind oder irreparable Schäden angesammelt haben.„Eps8 ist ein Protein mit vielen verschiedenen Funktionen, und es gibt noch viel zu erforschen“, sagte Manor.Zukünftige Forschungen umfassen die Untersuchung der Wirkung der Eps8-Gentherapie auf die Wiederherstellung des Hörvermögens in verschiedenen Entwicklungsstadien und ob es möglich ist, die Behandlungsmöglichkeiten zu verlängern.Zufällig veröffentlichte Professorin KarenB Avraham von der Universität Tel Aviv in Israel im November 2020 in der Zeitschrift EMBO Molecular Medicine [3] ihre Ergebnisse, in denen sie mithilfe einer innovativen Gentherapietechnologie ein harmloses synthetisches Adeno-assoziiertes Virus AAV9-PHP erzeugte.B: Der Gendefekt in den Haarzellen von Syne4-/- Mäusen wurde repariert, indem ein Virus, das die kodierende Sequenz von Syne4 trug, in das Innenohr von Mäusen injiziert wurde, wodurch es in die Haarzellen eindringen und das getragene genetische Material freisetzen konnte, wodurch sie reifen und normal funktionieren konnten (wie in Abb. 2).
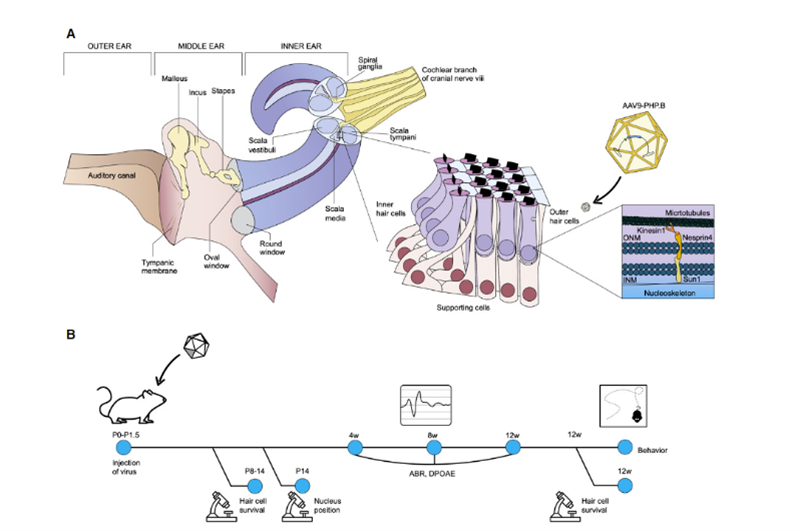
Abb. 2.Schematische Darstellung der Anatomie des Innenohrs mit Schwerpunkt auf dem Corti-Organ und der zellulären Funktion von Nesprin-4.
Es ist ersichtlich, dass der Einsatz der Gentherapie zur Behandlung von Erbkrankheiten auf Genebene durch Einfügen, Entfernen oder Korrigieren mutierter Gene zur Behandlung (d. h. Kontrolle der genetischen Veränderungen der Krankheit) eine hohe klinische Wirkung hat.Bewerbungsaussichten.Die aktuellen gentherapeutischen Methoden bei genetisch bedingter Taubheit lassen sich in folgende Kategorien einteilen:
Genersatz
Der Genersatz ist wohl die „einfachste“ Form der Gentherapie und basiert auf der Identifizierung und dem Ersatz eines defekten Gens durch eine normale oder Wildtyp-Kopie des Gens.Erste erfolgreiche Studie zur Innenohr-Gentherapie bei Hörverlust, der durch die Deletion des vesikulären Glutamattransporter-3-Gens (VGLUT3) verursacht wird;Die durch AAV1 vermittelte Abgabe einer exogenen VGLUT3-Überexpression in Innenohr-Haarzellen (IHCs) kann zu einer anhaltenden Wiederherstellung des Hörvermögens, einer teilweisen Wiederherstellung der synaptischen Bandmorphologie und konvulsiven Reaktionen führen [4].In den Beispielen, die die beiden in der Einleitung oben beschriebenen AAV-vermittelten Genaustausche umfassen, ist es jedoch wichtig zu beachten, dass sich die Mausmodelle, die für bestimmte Arten von erblichen Hörverluststörungen durch Genlöschung verwendet werden, zeitlich von Menschen unterscheiden und sich bei P1-Mäusen das Innenohr im reifen Entwicklungsstadium befindet.Im Gegensatz dazu wird der Mensch mit einem ausgereiften Innenohr geboren.Dieser Unterschied verhindert eine mögliche Anwendung der Mausergebnisse zur Behandlung erblicher Taubheitsstörungen beim Menschen, es sei denn, die Gentherapie wird an reifen Mäuseohren verabreicht.
Genbearbeitung: CRISPR/Cas9
Im Vergleich zum „Genersatz“ hat die Entwicklung der Gen-Editing-Technologie den Beginn der Behandlung genetischer Krankheiten an der Wurzel gebracht.Wichtig ist, dass die Gen-Editing-Methode die Mängel traditioneller Überexpressions-Gentherapiemethoden ausgleicht, die nicht für dominante erbliche Taubheitskrankheiten geeignet sind, und das Problem, dass die Überexpressionsmethode nicht lange anhält.Nachdem chinesische Forscher das mutierte Myo6C442Y-Allel in Myo6WT/C442Y-Mäusen mithilfe des AAV-SaCas9-KKH-Myo6-g2-Geneditierungssystems gezielt ausgeschaltet hatten, wurde die Hörfunktion des Modells innerhalb von 5 Monaten nach dem Knockout wiederhergestellt;Gleichzeitig wurde auch beobachtet, dass sich die Überlebensrate der Haarzellen im Innenohr verbesserte, die Form der Flimmerhärchen regelmäßiger wurde und die elektrophysiologischen Indikatoren korrigiert wurden [5].Dies ist weltweit die erste Studie, die die CRISPR/Cas9-Technologie zur Behandlung von erblicher Taubheit verwendet, die durch die Mutation des Myo6-Gens verursacht wird, und es handelt sich um einen wichtigen Forschungsfortschritt der Gen-Editing-Technologie zur Behandlung von erblicher Taubheit.Die klinische Übersetzung der Behandlung bietet eine solide wissenschaftliche Grundlage.
Methoden zur Verabreichung von Gentherapien
Damit die Gentherapie erfolgreich ist, können nackte DNA-Moleküle aufgrund ihrer Hydrophilie und der negativen Ladung der Phosphatgruppen nicht effektiv in Zellen eindringen. Um die Integrität der ergänzten Nukleinsäuremoleküle sicherzustellen, muss eine sichere und wirksame Methode ausgewählt werden.Die ergänzte DNA wird an die Zielzelle oder das Zielgewebe abgegeben.AAV wird aufgrund seiner hohen infektiösen Wirkung, geringen Immunogenität und seines breiten Tropismus für verschiedene Gewebetypen häufig als Transportvehikel zur Behandlung von Krankheiten eingesetzt.Gegenwärtig hat eine umfangreiche Forschungsarbeit den Tropismus verschiedener Subtypen von AAV im Verhältnis zu verschiedenen Zelltypen in der Cochlea der Maus bestimmt.Durch die Verwendung von AAV-Abgabeeigenschaften in Kombination mit zellspezifischen Promotoren kann eine zellspezifische Expression erreicht werden, wodurch Off-Target-Effekte reduziert werden können.Darüber hinaus werden als Alternative zu herkömmlichen AAV-Vektoren ständig neue synthetische AAV-Vektoren entwickelt, die eine überlegene Transduktionsfähigkeit im Innenohr aufweisen, wobei AAV2/Anc80L65 am häufigsten verwendet wird.Nicht-virale Verabreichungsmethoden können weiter in physikalische Methoden (Mikroinjektion und Elektroporation) und chemische Methoden (Lipid-basierte, polymerbasierte und Gold-Nanopartikel) unterteilt werden.Beide Ansätze wurden bei der Behandlung erblicher Taubheitsstörungen eingesetzt und zeigten unterschiedliche Vorteile und Einschränkungen.Zusätzlich zum Abgabevehikel für die Gentherapie als Vehikel können unterschiedliche Ansätze für die In-vivo-Genverabreichung basierend auf unterschiedlichen Zielzelltypen, Verabreichungswegen und therapeutischer Wirksamkeit eingesetzt werden.Aufgrund der komplizierten Struktur des Innenohrs ist es schwierig, Zielzellen zu erreichen, und die Verteilung von Genome-Editing-Mitteln erfolgt langsam.Das häutige Labyrinth befindet sich im knöchernen Labyrinth des Schläfenbeins und umfasst den Ductus cochlearis, den Ductus semicircularis, den Utriculus und den Ballon.Seine relative Isolation, die minimale Lymphzirkulation und die Trennung vom Blut durch eine Blutlabyrinth-Barriere schränken die wirksame systemische Verabreichung von Therapeutika nur an neugeborene Mäuse ein.Um für die Gentherapie geeignete Virustiter zu erhalten, ist eine direkte lokale Injektion viraler Vektoren in das Innenohr erforderlich.Zu den etablierten Injektionswegen gehören [6]: (1) runde Fenstermembran (RWM), (2) Tracheotomie, (3) endolymphatische oder perilymphatische Cochleostomie, (4) runde Fenstermembran plus Tubenfensterung (CF) (wie in Abb. 3).
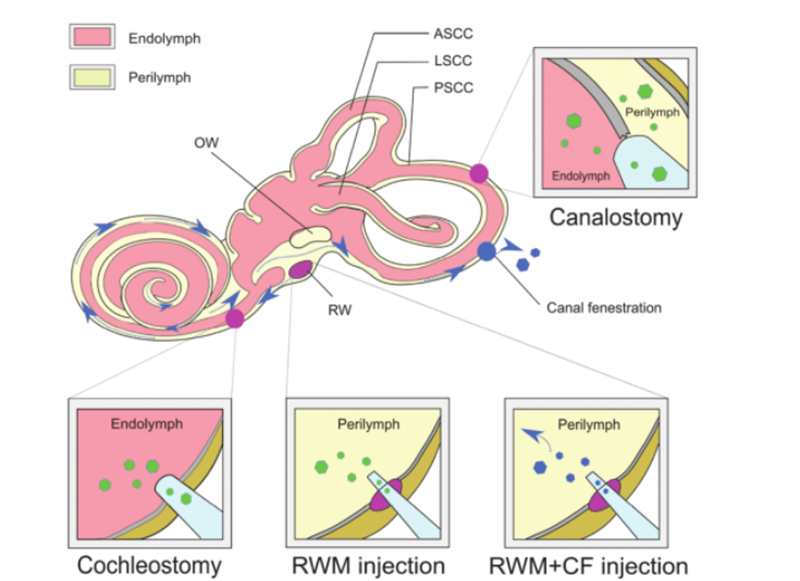
Abb. 3.Verabreichung einer Gentherapie an das Innenohr.
Obwohl in der Gentherapie auf der Grundlage klinisch-translationaler Ziele viele Fortschritte erzielt wurden, muss noch mehr Arbeit geleistet werden, bevor die Gentherapie zu einer Erstbehandlungsoption für Patienten mit genetisch bedingten Erkrankungen werden kann, insbesondere bei der Entwicklung sicherer und wirksamer Vektoren und Verabreichungsmethoden.Wir glauben jedoch, dass diese Art von Behandlungen in naher Zukunft zu einem festen Bestandteil der personalisierten Therapie werden und einen äußerst positiven Einfluss auf das Leben von Menschen mit genetischen Störungen und ihren Familien haben werden.
Foregene hat außerdem ein Hochdurchsatz-Screening-Kit für Zielgene auf den Markt gebracht, das schnell ist und reverse Transkription und qPCR-Reaktionen ohne RNA-Extraktion durchführen kann.
Produktlinks
Cell Direct RT-qPCR-Kit – Taqman/SYBR GREEN I
Für weitere Produktinformationen wenden Sie sich bitte an:
Zeitpunkt der Veröffentlichung: 02.09.2022



