



PCR (Polymerase-Kettenreaktion) ist eine der In-vitro-DNA-Amplifikationstechnologien mit einer mehr als 30-jährigen Geschichte.
Die PCR-Technologie wurde 1983 von Kary Mullis aus Cetus, USA, entwickelt. Mullis meldete 1985 ein PCR-Patent an und veröffentlichte im selben Jahr die erste wissenschaftliche PCR-Arbeit zum Thema Wissenschaft.Für seine Arbeit wurde Mullis 1993 mit dem Nobelpreis für Chemie ausgezeichnet.
Grundprinzipien der PCR
PCR kann Ziel-DNA-Fragmente um mehr als eine Million Mal verstärken.Das Prinzip beruht auf der Katalyse der DNA-Polymerase, wobei die DNA des Elternstrangs als Matrize und ein spezifischer Primer als Ausgangspunkt für die Verlängerung verwendet werden.Es wird in vitro durch Schritte wie Denaturierung, Annealing und Extension repliziert.Der Prozess der Tochterstrang-DNA, die zur Mutterstrang-Matrizen-DNA komplementär ist.

Der Standard-PCR-Prozess ist in drei Schritte unterteilt:
1.Denaturierung: Verwenden Sie hohe Temperaturen, um DNA-Doppelstränge zu trennen.Die Wasserstoffbindung zwischen DNA-Doppelsträngen wird bei hoher Temperatur (93-98℃) aufgebrochen.
2.Annealing: Nachdem die doppelsträngige DNA getrennt wurde, senken Sie die Temperatur, damit der Primer an die einzelsträngige DNA binden kann.
3.Verlängerung: Die DNA-Polymerase beginnt mit der Synthese komplementärer Stränge entlang der DNA-Stränge aus den gebundenen Primern, wenn die Temperatur gesenkt wird.Wenn die Verlängerung abgeschlossen ist, ist ein Zyklus abgeschlossen und die Anzahl der DNA-Fragmente verdoppelt sich
Durch 25- bis 35-maliges Durchführen dieser drei Schritte erhöht sich die Anzahl der DNA-Fragmente exponentiell.
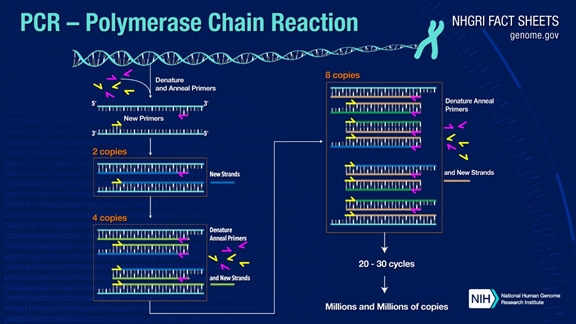
Der Einfallsreichtum der PCR besteht darin, dass unterschiedliche Primer für unterschiedliche Zielgene entwickelt werden können, sodass Zielgenfragmente in kurzer Zeit amplifiziert werden können.
Bisher lässt sich die PCR in drei Kategorien einteilen, nämlich gewöhnliche PCR, fluoreszierende quantitative PCR und digitale PCR.
Die erste Generation der gewöhnlichen PCR
Verwenden Sie ein gewöhnliches PCR-Amplifikationsgerät, um das Zielgen zu amplifizieren, und verwenden Sie dann eine Agarosegelelektrophorese, um das Produkt nachzuweisen. Es kann nur eine qualitative Analyse durchgeführt werden.
Die Hauptnachteile der PCR der ersten Generation:
1.Anfällig für unspezifische Amplifikation und falsch positive Ergebnisse.
2. Die Erkennung dauert lange und die Bedienung ist umständlich.
3. Es können nur qualitative Tests durchgeführt werden
Echtzeit-PCR der zweiten Generation
Echtzeit-PCR, auch qPCR genannt, verwendet fluoreszierende Sonden, die den Fortschritt des Reaktionssystems anzeigen können, überwacht die Akkumulation amplifizierter Produkte durch die Akkumulation von Fluoreszenzsignalen und beurteilt die Ergebnisse anhand der Fluoreszenzkurve.Sie kann mithilfe des Cq-Werts und der Standardkurve quantifiziert werden.
Da die qPCR-Technologie in einem geschlossenen System durchgeführt wird, verringert sich die Wahrscheinlichkeit einer Kontamination und das Fluoreszenzsignal kann für den quantitativen Nachweis überwacht werden. Daher ist sie in der klinischen Praxis am weitesten verbreitet und hat sich zur dominierenden Technologie in der PCR entwickelt.
Die in der quantitativen Echtzeit-Fluoreszenz-PCR verwendeten fluoreszierenden Substanzen können unterteilt werden in: TaqMan-Fluoreszenzsonde, Molecular Beacons und Fluoreszenzfarbstoff.
1)TaqMan-Fluoreszenzsonde:
Während der PCR-Amplifikation wird eine spezifische Fluoreszenzsonde hinzugefügt, während gleichzeitig ein Primerpaar hinzugefügt wird.Die Sonde ist ein Oligonukleotid und beide Enden sind mit einer Reporter-Fluoreszenzgruppe und einer Quencher-Fluoreszenzgruppe markiert.
Wenn die Sonde intakt ist, wird das von der Reportergruppe emittierte Fluoreszenzsignal von der Löschgruppe absorbiert;Während der PCR-Amplifikation spaltet und zersetzt die 5′-3′-Exonukleaseaktivität des Taq-Enzyms die Sonde, wodurch die Reporter-Fluoreszenzgruppe und der Quencher entstehen. Die Fluoreszenzgruppe wird getrennt, so dass das Fluoreszenzüberwachungssystem das Fluoreszenzsignal empfangen kann, d.
2) SYBR-Fluoreszenzfarbstoff:
Im PCR-Reaktionssystem wird ein Überschuss an SYBR-Fluoreszenzfarbstoff zugesetzt.Nachdem der SYBR-Fluoreszenzfarbstoff unspezifisch in den DNA-Doppelstrang eingebaut wurde, sendet er ein Fluoreszenzsignal aus.Das nicht in die Kette eingebaute SYBR-Farbstoffmolekül emittiert kein Fluoreszenzsignal, wodurch sichergestellt wird, dass das Fluoreszenzsignal vollständig mit der Zunahme der PCR-Produkte synchronisiert ist.SYBR bindet nur an doppelsträngige DNA, sodass anhand der Schmelzkurve festgestellt werden kann, ob die PCR-Reaktion spezifisch ist.

3) Molekulares Leuchtfeuer:
Es handelt sich um eine doppelt markierte Stamm-Schleifen-Oligonukleotidsonde, die an den Enden 5 und 3 eine Haarnadelstruktur von etwa 8 Basen bildet.Die Nukleinsäuresequenzen an beiden Enden sind komplementär gepaart, wodurch die fluoreszierende Gruppe und die Löschgruppe eng beieinander liegen.Schließen, es wird keine Fluoreszenz erzeugt.

Nachdem das PCR-Produkt erzeugt wurde, wird während des Annealing-Prozesses der mittlere Teil des Molecular Beacon mit einer spezifischen DNA-Sequenz gepaart und das Fluoreszenzgen vom Quencher-Gen getrennt, um Fluoreszenz zu erzeugen.

Die Hauptnachteile der PCR der zweiten Generation:
Die Empfindlichkeit ist immer noch unzureichend und die Erkennung von Proben mit geringer Kopienzahl ist ungenau.
Es besteht ein Einfluss des Hintergrundwertes und das Ergebnis ist störanfällig.
Bei Anwesenheit von PCR-Inhibitoren im Reaktionssystem sind die Nachweisergebnisse störanfällig.
Digitale PCR der dritten Generation
Die digitale PCR (DigitalPCR, dPCR, Dig-PCR) berechnet die Kopienzahl der Zielsequenz durch Endpunktdetektion und kann eine genaue absolute quantitative Detektion ohne Verwendung interner Kontrollen und Standardkurven durchführen.
Die digitale PCR nutzt die Endpunkterkennung und ist nicht vom Ct-Wert (Zyklusschwelle) abhängig, sodass die digitale PCR-Reaktion weniger von der Amplifikationseffizienz beeinflusst wird und die Toleranz gegenüber PCR-Reaktionsinhibitoren bei hoher Genauigkeit und Reproduzierbarkeit verbessert wird.
Aufgrund der Eigenschaften hoher Empfindlichkeit und hoher Genauigkeit wird es nicht leicht durch PCR-Reaktionsinhibitoren beeinträchtigt und kann ohne Standardprodukte eine echte absolute Quantifizierung erreichen, was zu einem Forschungs- und Anwendungs-Hotspot geworden ist.
Entsprechend den unterschiedlichen Formen der Reaktionseinheit kann diese in drei Haupttypen unterteilt werden: Mikrofluidik-, Chip- und Tröpfchensysteme.
1) Mikrofluidische digitale PCR, mdPCR:
Basierend auf der Mikrofluidik-Technologie wird die DNA-Vorlage abgetrennt.Die Mikrofluidik-Technologie kann die Proben-Nanoaufwertung oder die Erzeugung kleinerer Tröpfchen realisieren, aber die Tröpfchen benötigen eine spezielle Adsorptionsmethode und werden dann mit dem PCR-Reaktionssystem kombiniert.mdPCR wurde nach und nach durch andere Methoden ersetzt.
2) Tröpfchenbasierte digitale PCR, ddPCR:
Verwenden Sie die Technologie zur Erzeugung von Wasser-in-Öl-Tröpfchen, um die Probe in Tröpfchen zu verarbeiten, und teilen Sie das Reaktionssystem, das Nukleinsäuremoleküle enthält, in Tausende von nanoskaligen Tröpfchen auf, von denen jedes nicht das nachzuweisende Nukleinsäure-Zielmolekül oder ein oder mehrere zu testende Nukleinsäure-Zielmoleküle enthält.
3) Chipbasierte digitale PCR, cdPCR:
Nutzen Sie die integrierte Fluid-Pathway-Technologie, um viele Mikroröhrchen und Mikrokavitäten auf Siliziumwafern oder Quarzglas zu gravieren, steuern Sie den Fluss der Lösung durch verschiedene Steuerventile und teilen Sie die Probenflüssigkeit in Nanometer gleicher Größe in die Reaktionsvertiefungen für die digitale PCR-Reaktion auf, um eine absolute Quantifizierung zu erreichen.
Die Hauptnachteile der PCR der dritten Generation:
Die Ausrüstung und Reagenzien sind teuer.
Die Anforderungen an die Vorlagenqualität sind hoch.Wenn die Vorlagemenge die Mikrosystemmenge übersteigt, ist eine Quantifizierung nicht möglich, und wenn sie zu klein ist, wird die Quantifizierungsgenauigkeit verringert.
Auch bei unspezifischer Amplifikation kann es zu falsch positiven Ergebnissen kommen.
Zeitpunkt der Veröffentlichung: 30. Juli 2021



