



RT-qPCR wird aus der gewöhnlichen PCR-Technologie entwickelt.Es fügt dem herkömmlichen PCR-Reaktionssystem fluoreszierende Chemikalien (fluoreszierende Farbstoffe oder fluoreszierende Sonden) hinzu und erkennt den PCR-Annealing- und Extension-Prozess in Echtzeit anhand ihrer unterschiedlichen Lumineszenzmechanismen.Änderungen des Fluoreszenzsignals im Medium werden verwendet, um das Ausmaß der Produktveränderung in jedem PCR-Zyklus zu berechnen.Derzeit sind die gängigsten Methoden die Fluoreszenzfarbstoffmethode und die Sondenmethode.
Fluoreszierende Farbstoffmethode:
Einige fluoreszierende Farbstoffe wie SYBR Green Ⅰ, PicoGreen, BEBO usw. emittieren selbst kein Licht, sondern emittieren Fluoreszenz, nachdem sie an die kleine Furche der dsDNA gebunden haben.Daher kann das Gerät zu Beginn der PCR-Reaktion das Fluoreszenzsignal nicht erkennen.Wenn die Reaktion zur Annealing-Extension- (zweistufige Methode) oder Extension-Stufe (dreistufige Methode) übergeht, werden zu diesem Zeitpunkt die Doppelstränge geöffnet und die neue DNA-Polymerase aktiviert. Während der Strangsynthese werden fluoreszierende Moleküle in der kleinen dsDNA-Furche kombiniert und emittieren Fluoreszenz.Mit zunehmender Anzahl der PCR-Zyklen verbinden sich immer mehr Farbstoffe mit dsDNA und auch das Fluoreszenzsignal wird kontinuierlich verstärkt.Nehmen Sie als Beispiel SYBR Green Ⅰ.
Sondenmethode:
Die Taqman-Sonde ist die am häufigsten verwendete Hydrolysesonde.Am 5′-Ende der Sonde befindet sich eine fluoreszierende Gruppe, normalerweise FAM.Die Sonde selbst ist eine zum Zielgen komplementäre Sequenz.Am 3′-Ende des Fluorophors befindet sich eine Fluoreszenzlöschgruppe.Nach dem Prinzip des Fluoreszenzresonanzenergietransfers (Förster-Resonanzenergietransfer, FRET) kann die Anregung des Donormoleküls die Fluoreszenz des Akzeptormoleküls induzieren, während die Fluoreszenzlöschgruppe (Donor-Fluoreszenzmolekül) und die löschende Fluoreszenzgruppe (Akzeptor-Fluoreszenzmolekül) überlappen und der Abstand sehr gering ist (7-10 nm).Daher emittiert die Reporter-Fluoreszenzgruppe zu Beginn der PCR-Reaktion keine Fluoreszenz, wenn die Sonde im System frei und intakt ist.Beim Annealing binden Primer und Sonde an die Matrize.Während der Verlängerungsphase synthetisiert die Polymerase kontinuierlich neue Ketten.DNA-Polymerase hat eine 5′-3′-Exonukleaseaktivität.Beim Erreichen der Sonde hydrolysiert die DNA-Polymerase die Sonde von der Matrize, trennt die Reporter-Fluoreszenzgruppe von der Quencher-Fluoreszenzgruppe und setzt das Fluoreszenzsignal frei.Da zwischen der Sonde und der Vorlage eine Eins-zu-eins-Beziehung besteht, ist die Sondenmethode der Farbstoffmethode hinsichtlich der Genauigkeit und Empfindlichkeit des Tests überlegen.
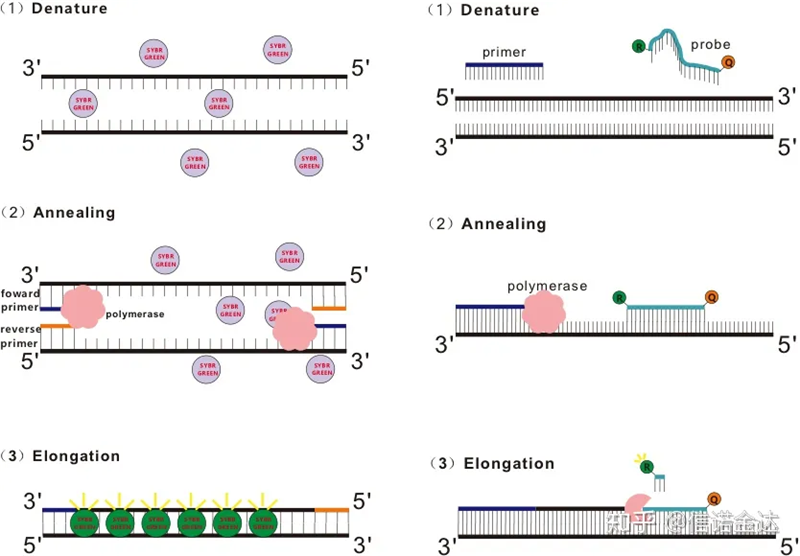
Abb. 1 Prinzip der qRT-PCR
Primer-Design
Grundsätze:
Die Primer sollten im konservierten Bereich der Nukleinsäurereihe konzipiert sein und Spezifität aufweisen.
Am besten verwenden Sie eine cDNA-Sequenz, aber auch eine mRNA-Sequenz ist akzeptabel.Wenn nicht, ermitteln Sie das Design der cds-Region der DNA-Sequenz.
Die Länge des fluoreszierenden quantitativen Produkts beträgt 80–150 bp, die längste beträgt 300 bp, die Primerlänge liegt im Allgemeinen zwischen 17–25 Basen und der Unterschied zwischen den Upstream- und Downstream-Primern sollte nicht zu groß sein.
Der G+C-Gehalt liegt zwischen 40 % und 60 %, am besten sind 45-55 %.
Der TM-Wert liegt zwischen 58-62 Grad.
Versuchen Sie, Primer-Dimere und Selbst-Dimere zu vermeiden (erscheinen nicht mehr als 4 Paare aufeinanderfolgender komplementärer Basen). Haarnadelstruktur, wenn unvermeidbar, stellen Sie ΔG < 4,5 kJ/mol ein -
Die spezifische Homologie der heterogen amplifizierten Sequenz beträgt vorzugsweise weniger als 70 % oder weist 8 komplementäre Basenhomologie auf.
Datenbank:
CottonFGD-Suche nach Schlüsselwörtern
Grundierungsdesign:
IDT-qPCR-Primerdesign
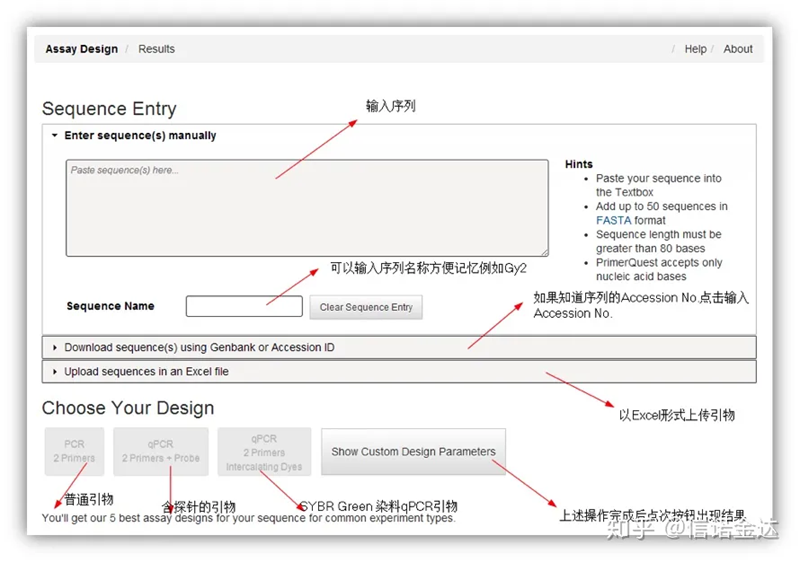
Abb. 2 IDT-Online-Primer-Design-Tool-Seite
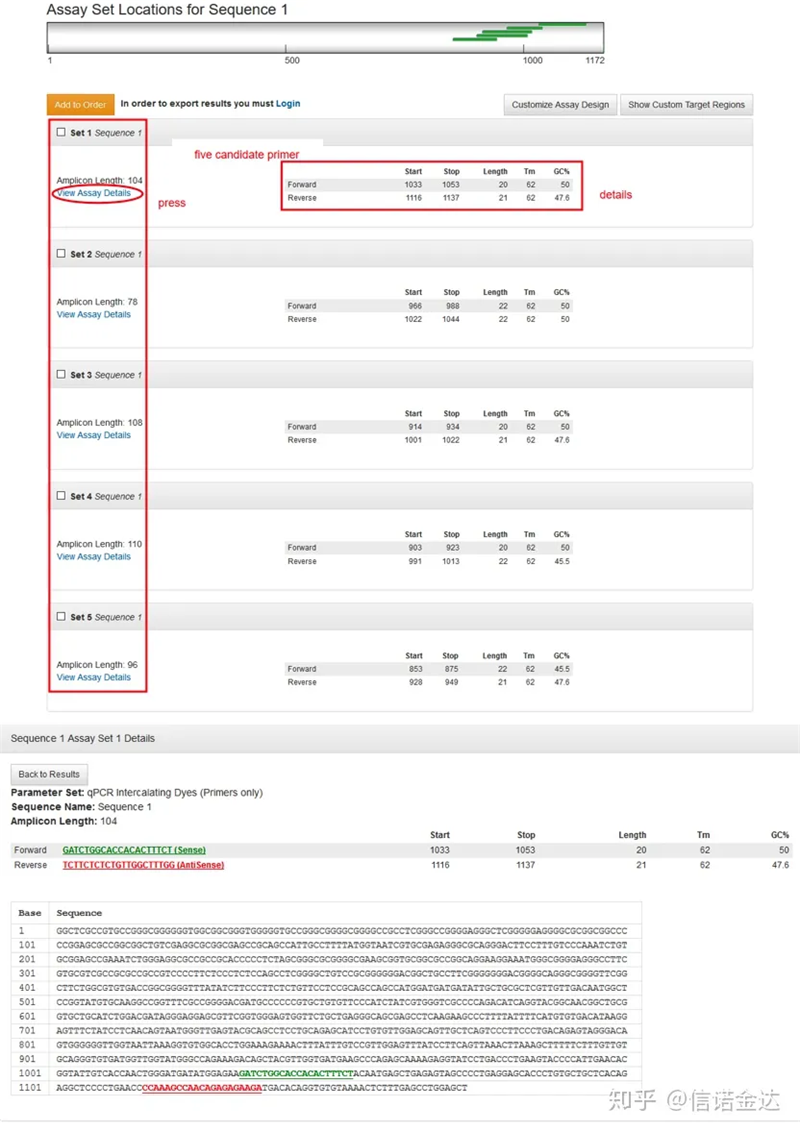
Abb. 3: Anzeige der Ergebnisseite
Design von lncRNA-Primern:
lncRNA:die gleichen Schritte wie mRNA.
miRNA:Das Prinzip der Stem-Loop-Methode: Da es sich bei allen miRNAs um kurze Sequenzen von etwa 23 nt handelt, kann kein direkter PCR-Nachweis durchgeführt werden, daher wird das Stem-Loop-Sequenz-Tool verwendet.Die Stamm-Schleifen-Sequenz ist eine einzelsträngige DNA von etwa 50 nt, die selbst eine Haarnadelstruktur bilden kann.3 'Das Ende kann als eine zum miRNA-Teilfragment komplementäre Sequenz gestaltet werden, dann kann die Ziel-miRNA während der reversen Transkription mit der Stamm-Loop-Sequenz verbunden werden, und die Gesamtlänge kann 70 bp erreichen, was mit der durch qPCR bestimmten Länge des amplifizierten Produkts übereinstimmt.Tailing-miRNA-Primer-Design.
Amplifikationsspezifischer Nachweis:
Online-Explosionsdatenbank: CottonFGD-Explosion nach Sequenzähnlichkeit
Lokaler Blast: Verwenden Sie Blast +, um einen lokalen Blast durchzuführen. Linux und MacOS können direkt eine lokale Datenbank erstellen. Das Win10-System kann auch nach der Installation von Ubuntu Bash durchgeführt werden.Erstellen Sie eine lokale Blast-Datenbank und einen lokalen Blast.Öffnen Sie Ubuntu Bash unter Win10.
Hinweis: Hochland-Baumwolle und Meeresinsel-Baumwolle sind tetraploide Nutzpflanzen, sodass das Ergebnis einer Explosion oft zwei oder mehr Übereinstimmungen sind.In der Vergangenheit war es wahrscheinlich, dass bei der Verwendung von NAU-CDs als Datenbank zur Durchführung von Explosionen zwei homologe Gene mit nur wenigen SNP-Unterschieden gefunden wurden.Normalerweise können die beiden homologen Gene nicht durch Primerdesign getrennt werden, daher werden sie als gleich behandelt.Wenn ein offensichtliches Indel vorhanden ist, wird der Primer normalerweise auf dem Indel entworfen, dies kann jedoch dazu führen, dass die Sekundärstruktur des Primers zunimmt. Die freie Energie wird höher, was zu einer Verringerung der Amplifikationseffizienz führt, was jedoch unvermeidbar ist.
Nachweis der Primer-Sekundärstruktur:
Schritte:Oligo 7 öffnen → Vorlagensequenz eingeben → Unterfenster schließen → Speichern → Primer auf Vorlage lokalisieren, Strg+D drücken, um die Primerlänge festzulegen → verschiedene Sekundärstrukturen analysieren, wie Selbstdimerisierungskörper, Heterodimer, Haarnadel, Fehlpaarung usw. Die letzten beiden Bilder in Abbildung 4 sind die Testergebnisse der Primer.Das Ergebnis des vorderen Primers ist gut, es gibt keine offensichtliche Dimer- und Haarnadelstruktur, keine kontinuierlichen komplementären Basen und der absolute Wert der freien Energie beträgt weniger als 4,5, während der hintere Primer kontinuierliche 6 Basen zeigt, die komplementär sind und die freie Energie 8,8 beträgt;Darüber hinaus erscheint am 3′-Ende ein schwerwiegenderes Dimer und ein Dimer mit 4 aufeinanderfolgenden Basen.Obwohl die freie Energie nicht hoch ist, kann das 3′-Dimer Chl die Amplifikationsspezifität und -effizienz ernsthaft beeinträchtigen.Darüber hinaus ist eine Überprüfung auf Haarnadeln, Heterodimere und Fehlpaarungen erforderlich.
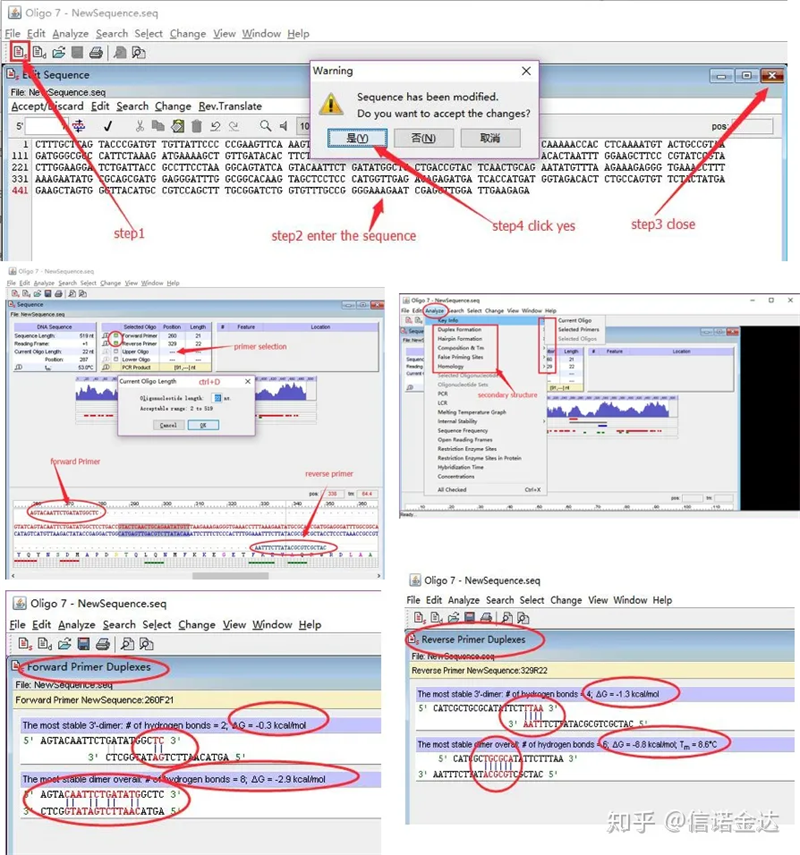
Abb. 3 Oligo7-Erkennungsergebnisse
Erkennung der Verstärkungseffizienz:
Die Amplifikationseffizienz der PCR-Reaktion beeinflusst die PCR-Ergebnisse erheblich.Auch bei der qRT-PCR ist die Amplifikationseffizienz besonders wichtig für die quantitativen Ergebnisse.Entfernen Sie andere Substanzen, Maschinen und Protokolle aus dem Reaktionspuffer.Auch die Qualität der Primer hat großen Einfluss auf die Amplifikationseffizienz der qRT-PCR.Um die Genauigkeit der Ergebnisse sicherzustellen, müssen sowohl die relative Fluoreszenzquantifizierung als auch die absolute Fluoreszenzquantifizierung die Amplifikationseffizienz der Primer erfassen.Es wird anerkannt, dass die effektive Effizienz der qRT-PCR-Amplifikation zwischen 85 % und 115 % liegt.Es gibt zwei Methoden:
1. Standardkurvenmethode:
A.cDNA mischen
B.Gradientenverdünnung
c.qPCR
D.Lineare Regressionsgleichung zur Berechnung der Verstärkungseffizienz
2. LinRegPCR
LinRegPCR ist ein Programm zur Analyse von Echtzeit-RT-PCR-Daten, auch quantitative PCR-Daten (qPCR) genannt, basierend auf SYBR Green oder einer ähnlichen Chemie.Das Programm verwendet nicht grundlinienkorrigierte Daten, führt eine Grundlinienkorrektur für jede Probe separat durch, bestimmt ein Fenster der Linearität und verwendet dann eine lineare Regressionsanalyse, um eine gerade Linie durch den PCR-Datensatz zu passen.Aus der Steigung dieser Linie wird die PCR-Effizienz jeder einzelnen Probe berechnet.Die mittlere PCR-Effizienz pro Amplikon und der Ct-Wert pro Probe werden zur Berechnung einer Startkonzentration pro Probe verwendet, ausgedrückt in willkürlichen Fluoreszenzeinheiten.Die Dateneingabe und -ausgabe erfolgt über eine Excel-Tabelle.Nur Probe
Mischen ist erforderlich, kein Gradient
Schritte sind erforderlich:(Nehmen Sie als Beispiel den Bole CFX96, nicht ganz eine Maschine mit klarem ABI)
Experiment:Es handelt sich um ein Standard-qPCR-Experiment.
qPCR-Datenausgabe:LinRegPCR kann zwei Formen von Ausgabedateien erkennen: RDML oder Quantifizierungs-Amplifikationsergebnis.Tatsächlich handelt es sich um den Echtzeit-Erkennungswert der Zyklusnummer und des Fluoreszenzsignals durch die Maschine, und die Verstärkung wird durch Analyse des Fluoreszenzänderungswerts der Effizienz des linearen Segments erhalten.
Datenauswahl: Theoretisch sollte der RDML-Wert verwendbar sein.Es wird geschätzt, dass das Problem meines Computers darin besteht, dass die Software RDML nicht erkennen kann, sodass ich den Excel-Ausgabewert als Originaldaten habe.Es wird empfohlen, zunächst eine grobe Überprüfung der Daten durchzuführen, z. B. auf Fehler beim Hinzufügen von Proben usw. Die Punkte können in den Ausgabedaten gelöscht werden (natürlich können Sie sie nicht löschen, LinRegPCR ignoriert diese Punkte in der späteren Phase).
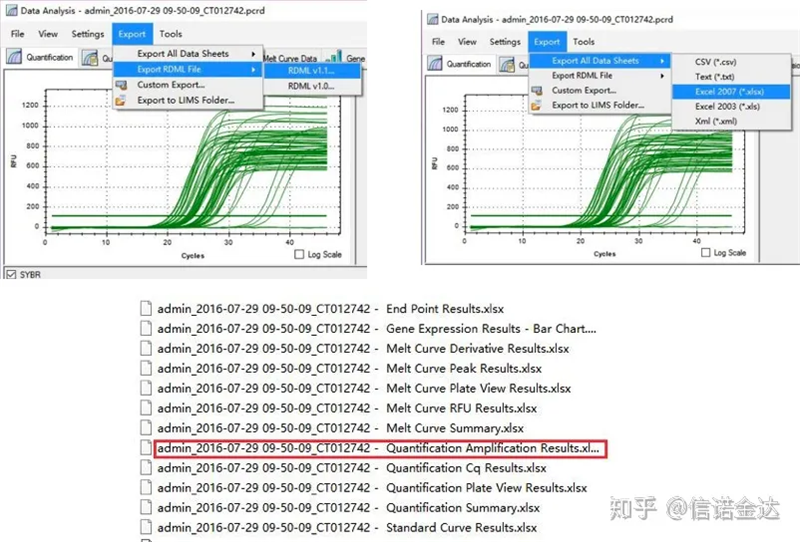
Abb. 5 qPCR-Datenexport
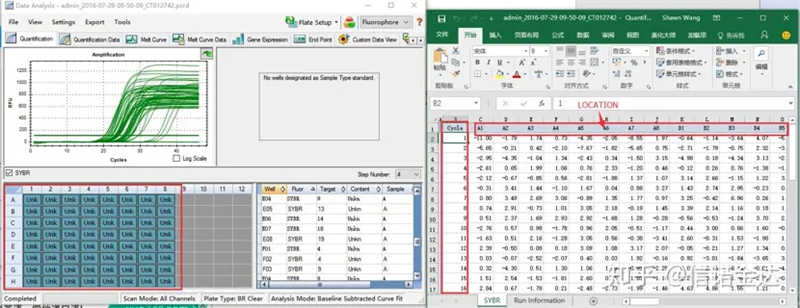
Abb. 6 Auswahl der Kandidatenproben
Dateneingabe:Öffnen Sie die Datei „Qualification Amplification Results.xls“, → öffnen Sie „LinRegPCR“ → „Datei“ → lesen Sie aus Excel → wählen Sie die Parameter aus, wie in Abbildung 7 dargestellt → „OK“ → klicken Sie auf „Basislinien ermitteln“.
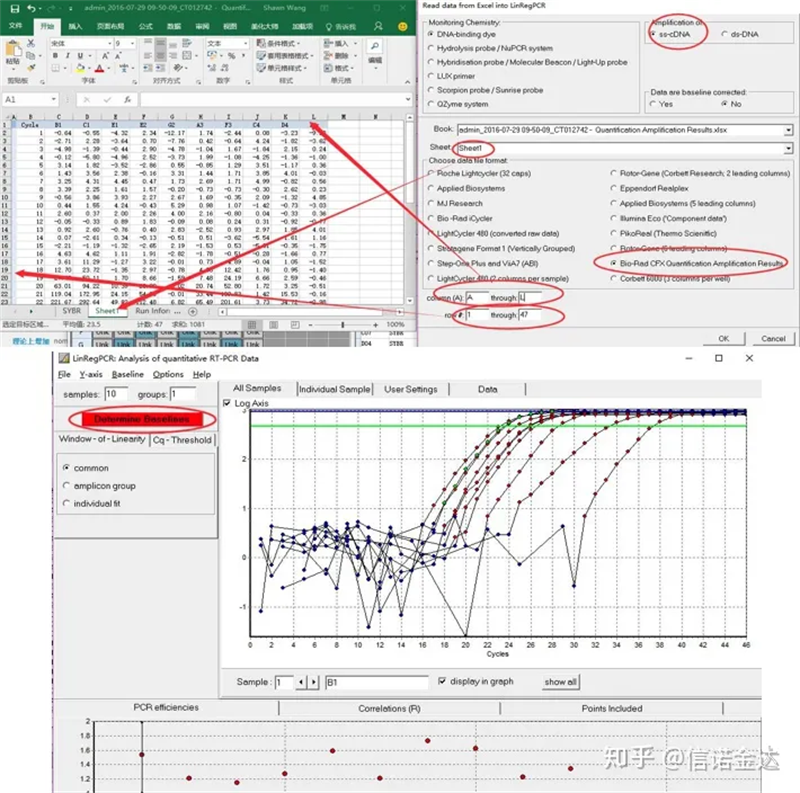
Abb. 7 Schritte der linRegPCR-Dateneingabe
Ergebnis:Wenn es keine Wiederholung gibt, ist keine Gruppierung erforderlich.Bei Wiederholungen kann die Gruppierung in der Probengruppierung bearbeitet werden, der Name des Gens wird in die Kennung eingetragen und dann wird das gleiche Gen automatisch gruppiert.Klicken Sie abschließend auf die Datei, exportieren Sie Excel und sehen Sie sich die Ergebnisse an.Die Amplifikationseffizienz und die R2-Ergebnisse jeder Vertiefung werden angezeigt.Zweitens wird bei der Aufteilung in Gruppen die korrigierte durchschnittliche Verstärkungseffizienz angezeigt.Stellen Sie sicher, dass die Amplifikationseffizienz jedes Primers zwischen 85 % und 115 % liegt.Wenn es zu groß oder zu klein ist, bedeutet dies, dass die Amplifikationseffizienz des Primers schlecht ist.
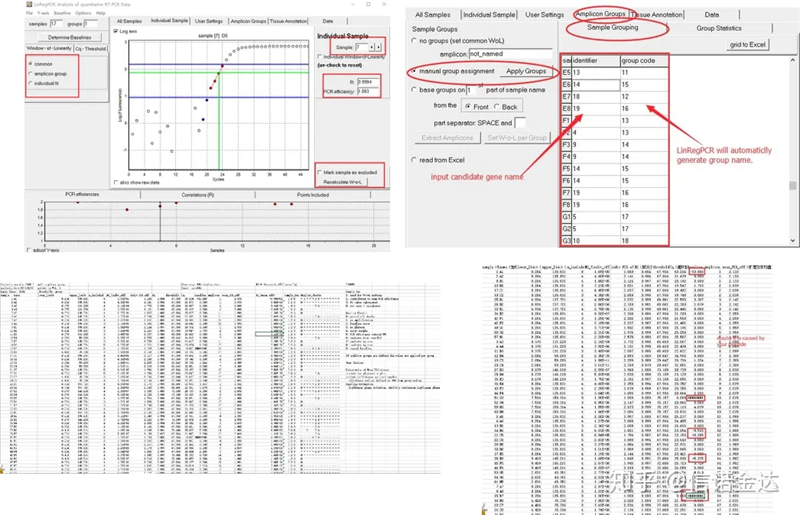
Abb. 8 Ergebnis und Datenausgabe
Experimenteller Ablauf:
Anforderungen an die RNA-Qualität:
Reinheit:1.72,0 weist darauf hin, dass möglicherweise restliches Isothiocyanat vorhanden ist.Saubere Nukleinsäure A260/A230 sollte etwa 2 betragen. Wenn eine starke Absorption bei 230 nm auftritt, weist dies darauf hin, dass organische Verbindungen wie Phenationen vorhanden sind.Darüber hinaus kann es durch 1,5 % Agarosegelelektrophorese nachgewiesen werden.Zeigen Sie auf den Marker, da die ssRNA keine Denaturierung aufweist und der Logarithmus des Molekulargewichts keine lineare Beziehung aufweist und das Molekulargewicht nicht korrekt ausgedrückt werden kann.Konzentration: Theoretischnichtweniger als 100 ng/ul, wenn die Konzentration zu niedrig ist, ist die Reinheit im Allgemeinen niedrig, nicht hoch
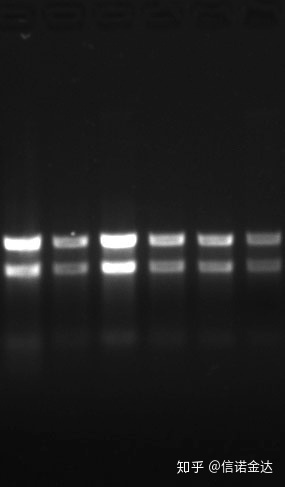
Abb.9 RNA-Gel
Wenn die Probe außerdem wertvoll und die RNA-Konzentration hoch ist, wird empfohlen, sie nach der Extraktion zu aliquotieren und die RNA für die reverse Transkription auf eine Endkonzentration von 100–300 ng/ul zu verdünnen.Inder Prozess der umgekehrten TranskriptionWenn mRNA transkribiert wird, werden Oligo (dt)-Primer, die spezifisch an PolyA-Schwänze binden können, für die reverse Transkription verwendet, während lncRNA und circRNA zufällige Hexamer-Primer (Random 6 mer) für die reverse Transkription der Gesamt-RNA verwenden. Für miRNA werden miRNA-spezifische Neck-Loop-Primer für die reverse Transkription verwendet.Viele Unternehmen haben mittlerweile spezielle Tailing-Kits auf den Markt gebracht.Für die Stem-Loop-Methode ist die Tailing-Methode bequemer, hat einen hohen Durchsatz und spart Reagenzien, aber die Wirkung der Unterscheidung von miRNAs derselben Familie sollte nicht so gut sein wie die Stem-Loop-Methode.Für jedes Reverse-Transkriptions-Kit gelten Anforderungen an die Konzentration genspezifischer Primer (Stamm-Loops).Die für miRNA verwendete interne Referenz ist U6.Bei der Stem-Loop-Inversion sollte ein Röhrchen U6 separat umgedreht und die vorderen und hinteren Primer von U6 direkt hinzugefügt werden.Sowohl circRNA als auch lncRNA können HKGs als interne Referenz verwenden.IncDNA-Nachweis,
Wenn es kein Problem mit der RNA gibt, sollte auch die cDNA in Ordnung sein.Wenn jedoch die Perfektion des Experiments angestrebt wird, ist es am besten, ein internes Referenzgen (Referenzgen, RG) zu verwenden, das gDNA von cds unterscheiden kann.Im Allgemeinen ist RG ein Housekeeping-Gen., HKG), wie in Abbildung 10 dargestellt;Zu dieser Zeit stellte ich Sojabohnen-Speicherprotein her und verwendete Actin7-haltige Introns als interne Referenz.Die Größe des amplifizierten Fragments dieses Primers in gDNA betrug 452 bp, und wenn cDNA als Matrize verwendet wurde, betrug sie 142 bp.Dann ergaben die Testergebnisse, dass ein Teil der cDNA tatsächlich durch gDNA kontaminiert war, und es wurde auch bewiesen, dass es kein Problem mit dem Ergebnis der reversen Transkription gab und sie als Vorlage für die PCR verwendet werden konnte.Es ist sinnlos, eine Agarose-Gelelektrophorese direkt mit cDNA durchzuführen, und es handelt sich um eine diffuse Bande, die nicht überzeugt.
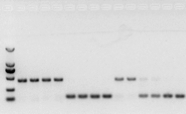
Abb. 10 cDNA-Nachweis
Die Bestimmung der qPCR-BedingungenGemäß dem Protokoll des Kits ist dies im Allgemeinen kein Problem, hauptsächlich im Schritt des tm-Werts.Wenn einige Primer während des Primerdesigns nicht gut entworfen wurden, was zu einem großen Unterschied zwischen dem tm-Wert und den theoretischen 60 °C führt, wird empfohlen, die cDNA nach dem Mischen der Proben mit Primern einer Gradienten-PCR durchzuführen und zu vermeiden, dass die Temperatur ohne Banden als TM-Wert eingestellt wird.
Datenanalyse
Die herkömmliche quantitative relative Fluoreszenz-PCR-Verarbeitungsmethode entspricht grundsätzlich 2-ΔΔCT.Datenverarbeitungsvorlage.
Verwandte Produkte:
Echtzeit-PCR einfachTM –Taqman
Echtzeit-PCR einfachTM –SYBR GRÜN I
RT Easy I (Master Premix für die Erststrang-cDNA-Synthese)
RT Easy II (Master Premix für die Erststrang-cDNA-Synthese für qPCR)
Zeitpunkt der Veröffentlichung: 14. März 2023



