



- PCR ist eine Methode zur Amplifikation von DNA aus einer kleinen Menge einer DNA-Matrize.RT-PCR nutzt die reverse Transkription, um aus einer RNA-Quelle eine DNA-Matrize zu erzeugen, die dann amplifiziert werden kann.
- PCR und RT-PCR sind typischerweise Endpunktreaktionen, während qPCR und RT-qPCR die Kinetik der Produktsyntheserate während der PCR-Reaktion nutzen, um die Menge der vorhandenen Vorlage zu quantifizieren.
- Neuere Methoden wie die digitale PCR ermöglichen eine absolute Quantifizierung der ursprünglichen DNA-Vorlage, während Methoden wie die isotherme PCR den Bedarf an teurer Ausrüstung reduzieren, um zuverlässige Ergebnisse zu liefern.
Die Polymerase-Kettenreaktion (PCR) ist eine relativ einfache und weit verbreitete molekularbiologische Technik zur Amplifikation und zum Nachweis von DNA- und RNA-Sequenzen.Im Vergleich zu herkömmlichen Methoden der DNA-Klonierung und -Amplifikation, die oft Tage dauern können, benötigt die PCR nur wenige Stunden.PCR ist hochempfindlich und erfordert nur eine minimale Vorlage für den Nachweis und die Amplifikation spezifischer Sequenzen.Grundlegende PCR-Methoden haben sich vom einfachen DNA- und RNA-Nachweis weiterentwickelt.Nachfolgend haben wir einen Überblick über die verschiedenen PCR-Methoden und die Reagenzien gegeben, die wir bei Enzo Life Sciences für Ihren Forschungsbedarf bereitstellen.Unser Ziel ist es, Wissenschaftlern dabei zu helfen, schnell auf PCR-Reagenzien zuzugreifen, die sie in ihrem nächsten Forschungsprojekt verwenden können!
PCR
Für die Standard-PCR benötigen Sie lediglich eine DNA-Polymerase, Magnesium, Nukleotide, Primer, die zu amplifizierende DNA-Vorlage und einen Thermocycler.Der PCR-Mechanismus ist so einfach wie sein Zweck: 1) doppelsträngige DNA (dsDNA) wird durch Hitze denaturiert, 2) Primer richten sich an den einzelnen DNA-Strängen aus und 3) die Primer werden durch DNA-Polymerase verlängert, was zu zwei Kopien davon führt ursprünglicher DNA-Strang.Der Denaturierungs-, Annealing- und Verlängerungsprozess über eine Reihe von Temperaturen und Zeiten wird als ein Amplifikationszyklus bezeichnet (Abb. 1).
|
|
| Abbildung 1.Schematische Darstellung eines Amplifikationszyklus durch PCR. |
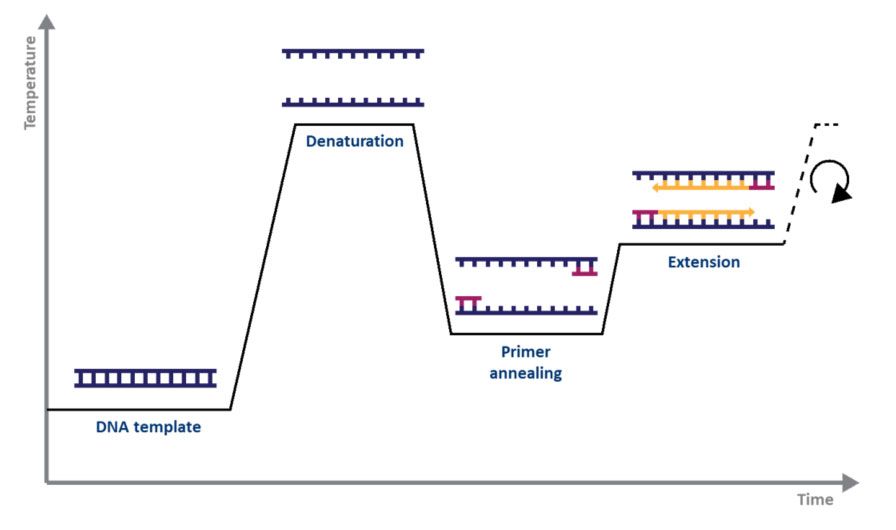
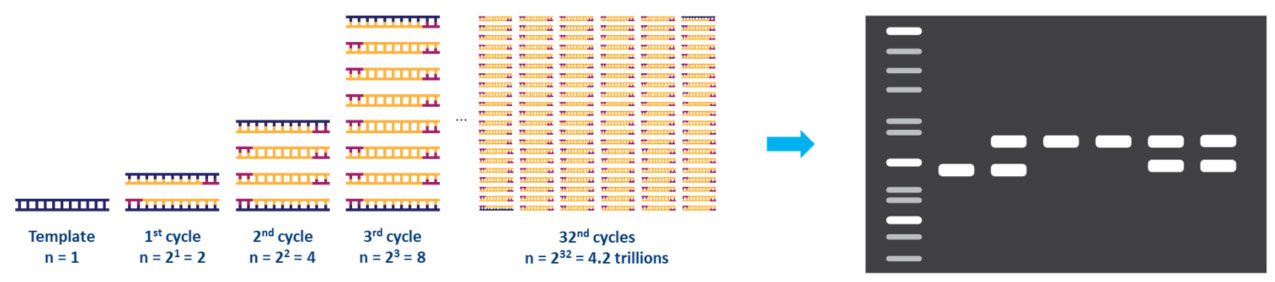
Jeder Schritt des Zyklus sollte für das verwendete Template und Primer-Set optimiert werden.Dieser Zyklus wird etwa 20–40 Mal wiederholt und das amplifizierte Produkt kann dann analysiert werden, typischerweise mittels Agarosegel (Abb. 2).
| |
| Figur 2.Amplifikation einer DNA-Matrize durch PCR und Analyse durch Agarosegelelektrophorese. |
Da es sich bei der PCR um eine hochempfindliche Methode handelt und für einzelne Reaktionen nur sehr geringe Volumina benötigt werden, empfiehlt sich die Herstellung eines Mastermixes für mehrere Reaktionen.Der Mastermix muss gut gemischt und dann nach der Anzahl der Reaktionen aufgeteilt werden, um sicherzustellen, dass jede Reaktion die gleiche Menge an Enzymen, dNTPs und Primern enthält.Viele Anbieter, wie zum Beispiel Enzo Life Sciences, bieten auch PCR-Mixe an, die bereits alles außer Primern und der DNA-Vorlage enthalten.
Guanin-/Cytosin-reiche (GC-reiche) Regionen stellen bei Standard-PCR-Techniken eine Herausforderung dar.GC-reiche Sequenzen sind stabiler als Sequenzen mit geringerem GC-Gehalt.Darüber hinaus neigen GC-reiche Sequenzen dazu, Sekundärstrukturen wie Haarnadelschleifen zu bilden.Daher ist es schwierig, GC-reiche Doppelstränge während der Denaturierungsphase vollständig zu trennen.Folglich kann die DNA-Polymerase den neuen Strang nicht ungehindert synthetisieren.Eine höhere Denaturierungstemperatur kann dies verbessern, und Anpassungen hin zu einer höheren Annealing-Temperatur und einer kürzeren Annealing-Zeit können die unspezifische Bindung von GC-reichen Primern verhindern.Zusätzliche Reagenzien können die Amplifikation GC-reicher Sequenzen verbessern.DMSO, Glycerin und Betain tragen dazu bei, die durch GC-Wechselwirkungen verursachten Sekundärstrukturen aufzubrechen und dadurch die Trennung der Doppelstränge zu erleichtern.
Hot-Start-PCR
Unspezifische Amplifikation ist ein Problem, das während der PCR auftreten kann.Die meisten DNA-Polymerasen, die für die PCR verwendet werden, funktionieren am besten bei Temperaturen um 68 °C bis 72 °C.Allerdings kann das Enzym auch bei niedrigeren Temperaturen aktiv sein, wenn auch in geringerem Maße.Bei Temperaturen weit unterhalb der Annealing-Temperatur können Primer unspezifisch binden und zu einer unspezifischen Amplifikation führen, selbst wenn die Reaktion auf Eis stattfindet.Dies kann durch den Einsatz von Polymerase-Inhibitoren verhindert werden, die sich erst ab einer bestimmten Temperatur von der DNA-Polymerase dissoziieren, daher der Begriff Hot-Start-PCR.Der Inhibitor kann ein Antikörper sein, der die Polymerase bindet und bei der anfänglichen Denaturierungstemperatur (typischerweise 95 °C) denaturiert.
High-Fidelity-Polymerase
Während DNA-Polymerasen ziemlich genau auf die ursprüngliche Matrizensequenz amplifizieren, können Fehler bei der Nukleotidzuordnung auftreten.Fehlpaarungen bei Anwendungen wie dem Klonen können zu verkürzten Transkripten und nachgeschalteten falsch übersetzten oder inaktiven Proteinen führen.Um diese Fehlpaarungen zu vermeiden, wurden Polymerasen mit „Korrekturlese“-Aktivität identifiziert und in den Arbeitsablauf integriert.Die erste Korrekturlese-Polymerase, Pfu, wurde 1991 in Pyrococcus furiosus identifiziert.Dieses Pfu-Enzym hat eine 3'- bis 5'-Exonukleaseaktivität.Während die DNA amplifiziert wird, entfernt die Exonuklease nicht übereinstimmende Nukleotide am 3'-Ende des Strangs.Anschließend wird das richtige Nukleotid ersetzt und die DNA-Synthese wird fortgesetzt.Die Identifizierung falscher Nukleotidsequenzen basiert auf der Bindungsaffinität des richtigen Nukleosidtriphosphats mit dem Enzym, wobei eine ineffiziente Bindung die Synthese verlangsamt und den korrekten Ersatz ermöglicht.Die Korrekturleseaktivität der Pfu-Polymerase führt im Vergleich zur Taq-DNA-Polymerase zu weniger Fehlern in der Endsequenz.In den letzten Jahren wurden weitere Korrekturleseenzyme identifiziert und Modifikationen des ursprünglichen Pfu-Enzyms vorgenommen, um die Fehlerrate bei der DNA-Amplifikation weiter zu reduzieren.
RT-PCR
Reverse Transkriptions-PCR oder RT-PCR ermöglicht die Verwendung von RNA als Vorlage.Ein zusätzlicher Schritt ermöglicht den Nachweis und die Amplifikation von RNA.Die RNA wird mithilfe der Reversen Transkriptase revers in komplementäre DNA (cDNA) transkribiert.Die Qualität und Reinheit der RNA-Vorlage sind entscheidend für den Erfolg der RT-PCR.Der erste Schritt der RT-PCR ist die Synthese eines DNA/RNA-Hybrids.Reverse Transkriptase verfügt außerdem über eine RNase H-Funktion, die den RNA-Anteil des Hybrids abbaut.Das einzelsträngige DNA-Molekül wird dann durch die DNA-abhängige DNA-Polymeraseaktivität der Reverse Transkriptase in cDNA vervollständigt.Die Effizienz der Erststrangreaktion kann den Amplifikationsprozess beeinflussen.Von hier an wird das Standard-PCR-Verfahren zur Amplifikation der cDNA verwendet.Die Möglichkeit, RNA mittels RT-PCR in cDNA umzuwandeln, hat viele Vorteile und wird vor allem für die Genexpressionsanalyse genutzt.RNA ist einzelsträngig und sehr instabil, was die Arbeit damit erschwert.Es dient üblicherweise als erster Schritt bei der qPCR, bei der RNA-Transkripte in einer biologischen Probe quantifiziert werden.
qPCR und RT-qPCR
Quantitative PCR (qPCR) wird zum Nachweis, zur Charakterisierung und zur Quantifizierung von Nukleinsäuren für zahlreiche Anwendungen eingesetzt.Bei der RT-qPCR werden RNA-Transkripte häufig quantifiziert, indem sie wie oben beschrieben zunächst revers in cDNA transkribiert und anschließend eine qPCR durchgeführt wird.Wie bei der Standard-PCR wird die DNA durch drei sich wiederholende Schritte amplifiziert: Denaturierung, Annealing und Verlängerung.Bei der qPCR ermöglicht die Fluoreszenzmarkierung jedoch die Erfassung von Daten im Verlauf der PCR.Aufgrund der Vielfalt der verfügbaren Methoden und Chemikalien bietet diese Technik viele Vorteile.
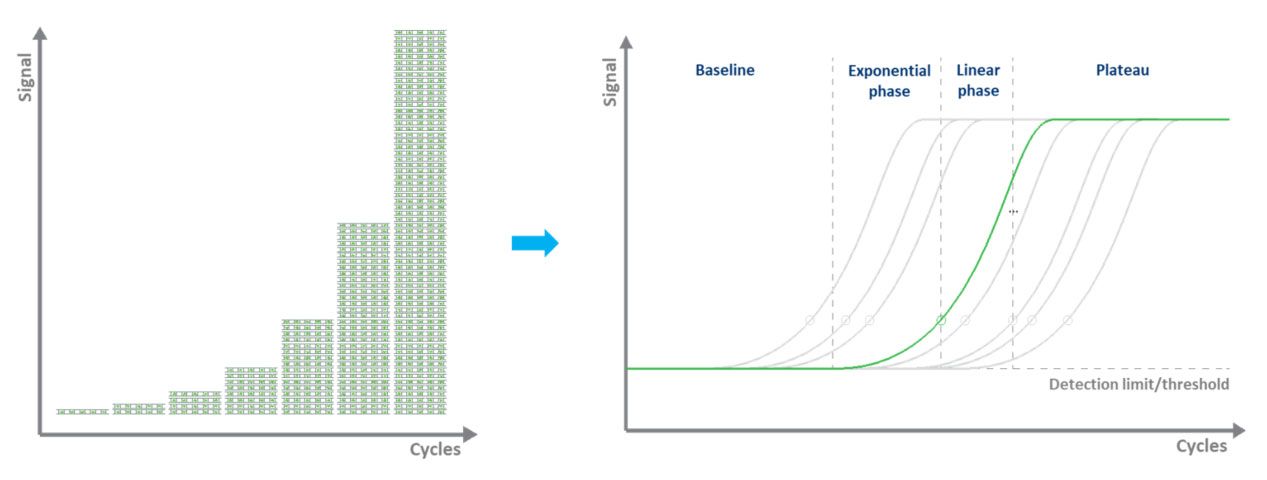
Bei der farbstoffbasierten qPCR (typischerweise grün) ermöglicht die Fluoreszenzmarkierung die Quantifizierung der amplifizierten DNA-Moleküle durch den Einsatz eines dsDNA-bindenden Farbstoffs.Während jedes Zyklus wird die Fluoreszenz gemessen.Das Fluoreszenzsignal steigt proportional zur Menge der replizierten DNA.Somit erfolgt die Quantifizierung der DNA in „Echtzeit“ (Abb. 3).Die Nachteile der farbstoffbasierten qPCR bestehen darin, dass jeweils nur ein Ziel untersucht werden kann und dass der Farbstoff an jegliche in der Probe vorhandene ds-DNA bindet.
|
|
| Figur 3.Amplifikation einer DNA-Vorlage durch qPCR und Messung des Fluoreszenzsignals in Echtzeit. |
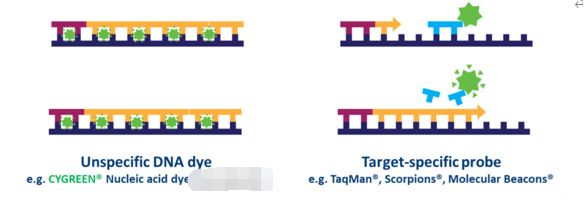
Bei der sondenbasierten qPCR können viele Ziele gleichzeitig in jeder Probe nachgewiesen werden. Dies erfordert jedoch die Optimierung und das Design einer oder mehrerer zielspezifischer Sonden, die zusätzlich zu den Primern verwendet werden.Es stehen verschiedene Arten von Sondendesigns zur Verfügung, der gebräuchlichste Typ ist jedoch eine Hydrolysesonde, die einen Fluorophor und einen Quencher enthält.Der Fluoreszenzresonanzenergietransfer (FRET) verhindert die Emission des Fluorophors über den Quencher, solange die Sonde intakt ist.Während der PCR-Reaktion wird die Sonde jedoch während der Primerverlängerung und Amplifikation der spezifischen Sequenz, an die sie gebunden ist, hydrolysiert.Die Abspaltung der Sonde trennt den Fluorophor vom Quencher und führt zu einem amplifikationsabhängigen Anstieg der Fluoreszenz (Abb. 4).Somit ist das Fluoreszenzsignal einer sondenbasierten qPCR-Reaktion proportional zur Menge der in der Probe vorhandenen Sondenzielsequenz.Da sondenbasierte qPCR spezifischer ist als farbstoffbasierte qPCR, wird diese Technologie häufig in qPCR-basierten diagnostischen Tests verwendet.
| |
| Figur 4.Unterschiede zwischen farbstoffbasierter und sondenbasierter qPCR. |
Isotherme Verstärkung
Die oben genannten PCR-Techniken erfordern teure Thermocycling-Geräte, um die Kammertemperaturen für die Denaturierungs-, Annealing- und Verlängerungsschritte genau zu erhöhen und zu senken.Es wurden eine Reihe von Techniken entwickelt, die keine derart präzisen Geräte erfordern und in einem einfachen Wasserbad oder sogar innerhalb der interessierenden Zellen durchgeführt werden können.Diese Techniken werden zusammenfassend als isotherme Verstärkung bezeichnet und basieren auf exponentieller, linearer oder kaskadenförmiger Verstärkung.
Die bekannteste Art der isothermen Verstärkung ist die schleifenvermittelte isotherme Verstärkung (LAMP).LAMP nutzt die exponentielle Amplifikation bei 65⁰C, um Template-DNA oder -RNA zu amplifizieren.Bei der Durchführung von LAMP werden vier bis sechs Primer, die zu Regionen der Ziel-DNA komplementär sind, mit einer DNA-Polymerase verwendet, um neue DNA zu synthetisieren.Zwei dieser Primer verfügen über komplementäre Sequenzen, die Sequenzen in den anderen Primern erkennen und sie binden, wodurch sich in der neu synthetisierten DNA eine „Schleifen“-Struktur bilden kann, die dann das Primer-Annealing in nachfolgenden Amplifikationsrunden unterstützt.LAMP kann mit mehreren Methoden sichtbar gemacht werden, einschließlich Fluoreszenz, Agarosegelelektrophorese oder Kolorimetrie.Die einfache Visualisierung und Erkennung des Vorhandenseins oder Nichtvorhandenseins eines Produkts durch Kolorimetrie und der Verzicht auf teure Geräte machten LAMP zu einer geeigneten Option für SARS-CoV-2-Tests in Bereichen, in denen klinische Labortests nicht ohne weiteres verfügbar waren, oder für die Lagerung und den Transport von Proben war nicht durchführbar, oder in Laboren, die bisher nicht über PCR-Thermocycling-Geräte verfügten.
Zeitpunkt der Veröffentlichung: 19. August 2023



