



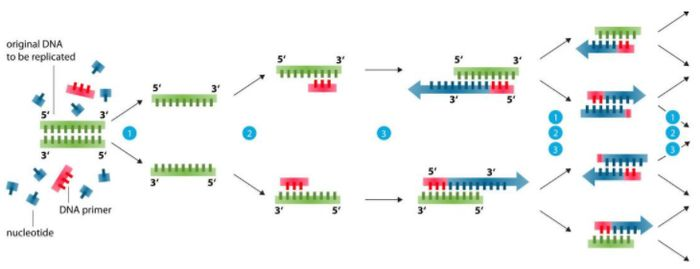
Primer-Designbasis (99 % der Probleme können gelöst werden)
1. Primerlänge: Das Lehrbuch erfordert 15-30 bp, normalerweise etwa 20 bp.Die tatsächliche Bedingung ist besser, 18-24 bp zu sein, um die Spezifität sicherzustellen, aber je länger, desto besser, ein zu langer Primer verringert auch die Spezifität und verringert die Ausbeute.
2. Primeramplifikationsspanne: 200–500 bp sind angemessen, und das Fragment kann unter bestimmten Bedingungen auf 10 kb erweitert werden.
3. Primerbasis: Der Gehalt an G+C sollte 40–60 % betragen. Zu wenig G+C-Verstärkungseffekt ist nicht gut, zu viel G+C kann leicht zu unspezifischen Banden führen.ATGC wird am besten zufällig verteilt, wobei Cluster von mehr als 5 Purin- oder Pyrimidinnukleotiden vermieden werden.Multi-GC für das 5‘-Ende und Zwischensequenzen zur Erhöhung der Stabilität, vermeiden Sie reichhaltige GC am 3‘-Ende, keine GC für die letzten 3 Basen oder keine GC für 3 der letzten 5 Basen.
4. Vermeiden Sie Sekundärstrukturen in Primern und vermeiden Sie die Komplementierung zwischen zwei Primern, insbesondere die Komplementierung am 3'-Ende, da sonst Primer-Dimere gebildet werden und unspezifische verstärkte Banden erzeugt werden.
5. Die Basen am 3'-Ende der Primer, insbesondere die letzte und vorletzte Base, sollten streng gepaart sein, um PCR-Fehler aufgrund ungepaarter terminaler Basen zu vermeiden.
6. Primer haben geeignete Spaltstellen oder können mit solchen hinzugefügt werden, und die amplifizierte Zielsequenz sollte vorzugsweise geeignete Spaltstellen haben, was für die Spaltungsanalyse oder das molekulare Klonen sehr vorteilhaft ist.
7. Spezifität der Primer: Primer sollten keine offensichtliche Homologie mit anderen Sequenzen in der Nukleinsäuresequenzdatenbank aufweisen.
8. Lernen Sie den Umgang mit Software: PP5, Oligo6, DNAstar, Vector NTI, Primer3 (Dieses Online-Design funktioniert am besten).
Der oben genannte Inhalt kann mindestens 99 % der Primer-Designprobleme lösen.
Kontrollieren Sie die Details des Primer-Designs
1. Primerlänge
Die allgemeine Primerlänge beträgt 18–30 Basen.Im Allgemeinen ist die Länge des Primers der wichtigste Faktor, der die Tempertemperatur des Primers bestimmt.Die Glühtemperatur des Primers wird im Allgemeinen ausgewählt (Tm-Wert -5℃), und einige verwenden den Tm-Wert direkt.Die folgenden Formeln können zur groben Berechnung der Annealing-Temperatur von Primern verwendet werden.
Wenn die Länge des Primers weniger als 20 bp beträgt: [4(G+C)+2(A+T)]-5℃
Wenn die Länge des Primers mehr als 20 bp beträgt: 62,3℃+0,41℃(%GC)-500/Länge-5℃
Darüber hinaus können viele Softwareprogramme auch zur Berechnung der Glühtemperatur verwendet werden. Das Berechnungsprinzip ist unterschiedlich, sodass der berechnete Wert manchmal eine kleine Lücke aufweist.Um PCR-Reaktionen zu optimieren, werden für beste Effizienz und Spezifität die kürzesten Primer verwendet, die Annealing-Temperaturen von nicht weniger als 54 °C gewährleisten.
Insgesamt erhöht sich die Primerspezifität mit jedem zusätzlichen Nukleotid um den Faktor vier, sodass die minimale Primerlänge für die meisten Anwendungen 18 Nukleotide beträgt.Die Obergrenze der Primerlänge ist nicht sehr wichtig, sondern hängt hauptsächlich mit der Reaktionseffizienz zusammen.Aufgrund der Entropie ist die Geschwindigkeit, mit der der Primer anlagert, umso geringer, je länger der Primer ist, um an die Ziel-DNA zu binden und eine stabile doppelsträngige Matrize für die Bindung der DNA-Polymerase zu bilden.
Bei der Verwendung von Software zum Entwerfen von Primern kann die Länge der Primer wiederum durch den TM-Wert bestimmt werden, insbesondere für Primer der quantitativen Fluoreszenz-PCR sollte TM=60℃ oder so kontrolliert werden.
2.GC-Inhalt
Im Allgemeinen beträgt der G+C-Gehalt in Primersequenzen 40–60 %, und der GC-Gehalt und der Tm-Wert eines Primerpaars sollten koordiniert werden.Wenn der Primer eine starke GC- oder AT-Tendenz aufweist, kann eine entsprechende Menge an A-, T- oder G- und C-Schwanz am 5'-Ende des Primers hinzugefügt werden.
3. Glühtemperatur
Die Glühtemperatur sollte 5℃ niedriger sein als die Entkettungstemperatur.Wenn die Anzahl der Primerbasen gering ist, kann die Annealing-Temperatur entsprechend erhöht werden, was die Spezifität der PCR erhöhen kann.Wenn die Anzahl der Basen groß ist, kann die Glühtemperatur entsprechend gesenkt werden.Der Annealing-Temperaturunterschied zwischen einem Primerpaar von 4℃ ~ 6℃ hat keinen Einfluss auf die PCR-Ausbeute, aber idealerweise ist die Annealing-Temperatur eines Primerpaares gleich und kann zwischen 55℃ ~ 75℃ variieren.
4. Vermeiden Sie den Sekundärstrukturbereich der Amplifikationsvorlage
Bei der Auswahl des amplifizierten Fragments ist es am besten, den Sekundärstrukturbereich der Vorlage zu meiden.Die stabile Sekundärstruktur des Zielfragments kann durch entsprechende Computersoftware vorhergesagt und geschätzt werden, was bei der Vorlagenauswahl hilfreich ist.Experimentelle Ergebnisse zeigen, dass die Erweiterung oft nicht erfolgreich ist, wenn die freie Energie (△G) der zu erweiternden Region weniger als 58,6 lkJ/mol beträgt.
5. Nichtübereinstimmung mit der Ziel-DNA
Wenn die amplifizierte Ziel-DNA-Sequenz groß ist, kann ein Primer an mehrere Teile der Ziel-DNA binden, was dazu führt, dass im Ergebnis mehrere Banden erscheinen.Diesmal ist es notwendig, die BLAST-Softwaretest-Website zu verwenden:http://www.ncbi.nlm.nih.gov/BLAST/.Wählen Sie Zwei Sequenzen ausrichten (bl2seq).
Das Einfügen von Primersequenzen in Zone 1 und Ziel-DNA-Sequenzen in Zone 2 ist austauschbar, und BLAST berechnet Komplementär-, Antisense- und andere Möglichkeiten, sodass Benutzer nicht beachten müssen, ob beide Ketten Sense-Ketten sind.Sie können die GI-Nummer auch eingeben, wenn Sie die GI-Nummer der Sequenz in der Datenbank kennen, sodass Sie keinen großen Abschnitt der Sequenz einfügen müssen.Klicken Sie abschließend auf „Ausrichten bei 3“, um zu sehen, ob der Primer mehrere homologe Stellen in der Ziel-DNA aufweist.
6. Primer-Terminal
Am 3'-Ende des Primers beginnt die Verlängerung, daher ist es wichtig zu verhindern, dass dort Fehlpaarungen beginnen.Das 3'-Ende sollte nicht mehr als 3 aufeinanderfolgende G oder C sein, da dies dazu führt, dass der Primer fälschlicherweise in der G+C-Anreicherungssequenzregion ausgelöst wird.Das 3'-Ende kann keine Sekundärstruktur bilden, außer bei speziellen PCR-Reaktionen (AS-PCR), bei denen das 3'-Ende des Primers nicht fehlgepaart sein darf.Wenn beispielsweise die kodierende Region amplifiziert wird, sollte das 3'-Ende des Primers nicht an der dritten Position des Codons enden, da die dritte Position des Codons zur Degeneration neigt, was sich auf die Spezifität und Effizienz der Amplifikation auswirkt.Beachten Sie bei der Verwendung von Annexionsprimern die Codon-Verwendungstabelle, achten Sie auf die biologische Präferenz, verwenden Sie keine Annexionsprimer am 3′-Ende und verwenden Sie eine höhere Primerkonzentration (1uM-3uM).
7. Sekundärstruktur von Primern
Die Primer selbst sollten keine komplementären Sequenzen aufweisen, da sich die Primer sonst selbst zu Haarnadelstrukturen falten und diese Sekundärstruktur aufgrund sterischer Hinderung die Bindung von Primern und Templates beeinträchtigt.Bei einer künstlichen Beurteilung sollten die fortlaufenden komplementären Basen der Primer selbst nicht größer als 3 bp sein.Es sollte keine Komplementarität zwischen den beiden Primern bestehen, insbesondere sollte die komplementäre Überlappung des 3'-Endes vermieden werden, um die Bildung von Primer-Dimeren zu verhindern.Im Allgemeinen sollte zwischen einem Primerpaar nicht mehr als 4 aufeinanderfolgende Basenhomologien oder -komplementaritäten vorliegen.
8. Fügen Sie Marker oder Loci hinzu
Das 5'-Ende hat kaum Einfluss auf die Amplifikationsspezifität und kann daher modifiziert werden, ohne die Amplifikationsspezifität zu beeinträchtigen.Die Modifikation des 5'-Endes des Primers umfasste: Hinzufügen einer Enzymrestriktionsstelle;Markiertes Biotin, Fluoreszenz, Digoxin, Eu3+ usw. Einführung proteinbindender DNA-Sequenzen;Einführung von Mutationsstellen, Einfügen und Fehlen von Mutationssequenzen und Einfügen von Promotorsequenzen usw. Die zusätzlichen Basen wirken sich mehr oder weniger auf die Effizienz der Amplifikation aus und erhöhen die Wahrscheinlichkeit der Bildung von Primerdimeren, für den nächsten Schritt müssen jedoch einige Zugeständnisse gemacht werden.Zusätzliche Sequenzen, die auf der Zielsequenz nicht vorhanden sind, wie etwa Restriktionsstellen und Promotorsequenzen, können an das 5′-Ende des Primers hinzugefügt werden, ohne die Spezifität zu beeinträchtigen.Diese Sequenzen werden nicht in die Berechnung der Primer-Tm-Werte einbezogen, sollten aber auf Komplementarität und interne Sekundärstruktur getestet werden.
9. Subklone
Meistens handelt es sich bei der PCR nur um eine vorläufige Klonierung, und dann müssen wir das Zielfragment in verschiedene Vektoren subklonieren, sodass wir zusätzliche Basen für den nächsten Vorgang im PCR-Schritt entwerfen müssen.
Einige für die Subklonierung konzipierte Sequenzen sind nachstehend zusammengefasst.
Restriktionsendonuklease-Restriktionsstelle wurde hinzugefügt
Das Hinzufügen von Enzymrestriktionsstellen ist die am häufigsten verwendete Methode zur Subklonierung von PCR-Produkten.Im Allgemeinen besteht die Spaltungsstelle aus sechs Basen. Zusätzlich zum 5'-Ende der Spaltungsstelle müssen 2 bis 3 Schutzbasen hinzugefügt werden.Allerdings ist die Anzahl der von verschiedenen Enzymen benötigten Schutzbasen unterschiedlich.Beispielsweise erfordert SalⅠ keine Schutzbasis, EcoRⅤ erfordert 1 Schutzbasis, NotⅠ erfordert 2 Schutzbasen und Hind Ⅲ erfordert 3 Schutzbasen.
LIC fügt den Schwanz hinzu
Der vollständige Name von LIC ist Ligation-Independent Cloning, eine Klonierungsmethode, die Navogen speziell für seinen Teil des pET-Vektors erfunden hat.Der mit der LIC-Methode hergestellte pET-Träger weist nichtkomplementäre 12–15 Basen lange Einzelstrang-Klebeenden auf, die die entsprechenden Klebeenden auf dem Ziel-Insert-Fragment ergänzen.Für Amplifikationszwecke sollte die Primer-5'-Sequenz des eingefügten Fragments den LIC-Vektor ergänzen.Die 3′→5′-Extranect-Aktivität der T4-DNA-Polymerase kann nach kurzer Zeit ein einzelsträngiges klebriges Ende auf dem eingefügten Fragment bilden.Da das Produkt nur durch gegenseitiges Annealing des vorbereiteten Insertfragments und des Vektors gebildet werden kann, ist diese Methode sehr schnell und effizient und es handelt sich um eine gerichtete Klonierung.
Gezielter TA-Klon fügt Schwanz hinzu
Das TA-Klonen war nicht in der Lage, das Fragment in einen Vektor zu lenken, daher führte Invitrogen später einen Vektor ein, der auf das Klonen abzielte und an einem Ende vier prominente Basen GTGGS enthielt.Daher sollten beim Design von PCR-Primern entsprechend komplementäre Sequenzen hinzugefügt werden, damit Fragmente „orientiert“ werden können.
Wenn Sie wenig Zeit haben, können Sie die direkte Synthese ausprobieren, indem Sie das Gen mit dem Vektor kombinieren, was wir in der Muskulatur als ET-Gensynthese bezeichnen.
D. In-Fusion-Klonmethode
Keine Ligase erforderlich, keine lange Reaktion erforderlich.Solange beim Design von Primern eine Sequenz an beiden Enden des linearisierten Vektors eingeführt wird, dann das PCR-Produkt und der linearisierte Vektor in die BSA enthaltende Infusionsenzymlösung gegeben und eine halbe Stunde lang bei Raumtemperatur gehalten werden, kann die Transformation durchgeführt werden.Diese Methode eignet sich besonders für die Umwandlung großer Mengen.
10. Primer zusammenführen
Manchmal sind nur begrenzte Sequenzinformationen über das Primerdesign bekannt.Wenn beispielsweise nur die Aminosäuresequenz bekannt ist, kann der Merging-Primer entworfen werden.Ein Merger-Primer ist eine Mischung verschiedener Sequenzen, die alle verschiedenen Basenmöglichkeiten darstellen, die eine einzelne Aminosäure kodieren.Um die Spezifität zu erhöhen, können Sie auf die Codon-Verwendungstabelle zurückgreifen, um die Annexion entsprechend den Basisverwendungspräferenzen verschiedener Organismen zu reduzieren.Hypoxanthin kann mit allen Basen kombiniert werden, um die Tempertemperatur des Primers zu senken.Verwenden Sie nicht die angehängten Basen am 3‘-Ende des Primers, da das Annealing der letzten 3 Basen am 3‘-Ende ausreicht, um die PCR an der falschen Stelle zu starten.Höhere Primerkonzentrationen (1 μM bis 3 μM) werden verwendet, da Primer in vielen Annexionsmischungen nicht spezifisch für die Zielvorlage sind.
PCR-RohstoffeKontrolle
1. Primermenge
Die Konzentration jedes Primers beträgt 0,1 ~ 1 umol oder 10 ~ 100 pmol.Es ist besser, das gewünschte Ergebnis mit der geringsten Grundierungsmenge zu erzielen.Eine hohe Primerkonzentration führt zu Fehlpaarungen und unspezifischer Amplifikation und erhöht die Wahrscheinlichkeit der Bildung von Dimeren zwischen Primern.
2. Primerkonzentration
Die Konzentration der Primer beeinflusst die Spezifität.Die optimale Primerkonzentration liegt im Allgemeinen zwischen 0,1 und 0,5 μM.Höhere Primerkonzentrationen führen zur Amplifikation unspezifischer Produkte.
3. Glühtemperatur des Primers
Ein weiterer wichtiger Parameter für Primer ist die Schmelztemperatur (Tm).Dies ist die Temperatur, bei der 50 % der Primer und Komplementärsequenzen als doppelsträngige DNA-Moleküle dargestellt werden.Tm ist erforderlich, um die PCR-Annealing-Temperatur einzustellen.Idealerweise ist die Annealing-Temperatur niedrig genug, um ein effektives Annealing der Primer mit der Zielsequenz zu gewährleisten, aber hoch genug, um unspezifische Bindungen zu reduzieren.Angemessene Glühtemperatur von 55℃ bis 70℃.Die Glühtemperatur wird im Allgemeinen 5 °C niedriger als die Tm des Primers eingestellt.
Für die Einstellung von Tm gibt es mehrere Formeln, die je nach verwendeter Formel und Primersequenz stark variieren.Da die meisten Formeln einen geschätzten Tm-Wert liefern, sind alle Glühtemperaturen nur ein Ausgangspunkt.Die Spezifität kann durch die Analyse mehrerer Reaktionen verbessert werden, die die Glühtemperatur schrittweise erhöhen.Beginnen Sie unterhalb der geschätzten Tm-5℃ und erhöhen Sie die Glühtemperatur schrittweise in Schritten von 2℃.Eine höhere Tempertemperatur verringert die Bildung von Primerdimeren und unspezifischen Produkten.Für beste Ergebnisse sollten die beiden Primer ungefähre Tm-Werte haben.Wenn der Tm-Unterschied der Primerpaare mehr als 5 °C beträgt, zeigen die Primer einen erheblichen Fehlstart, da im Zyklus eine niedrigere Annealing-Temperatur verwendet wird.Wenn die Tm der beiden Primer unterschiedlich sind, stellen Sie die Annealing-Temperatur auf 5 °C niedriger als die niedrigste Tm ein.Alternativ können zur Erhöhung der Spezifität zunächst fünf Zyklen bei Annealing-Temperaturen durchgeführt werden, die für eine höhere Tm ausgelegt sind, gefolgt von den restlichen Zyklen bei Annealing-Temperaturen, die für eine niedrigere Tm ausgelegt sind.Dadurch kann unter engen Bedingungen eine Teilkopie der Zielvorlage erhalten werden.
4. Reinheit und Stabilität des Primers
Die Standardreinheit kundenspezifischer Primer ist für die meisten PCR-Anwendungen ausreichend.Die Entfernung von Benzoyl- und Isobutylylgruppen durch Entsalzen ist minimal und beeinträchtigt daher die PCR nicht.Einige Anwendungen erfordern eine Reinigung, um alle nicht vollständigen Sequenzen im Syntheseprozess zu entfernen.Diese verkürzten Sequenzen treten auf, weil die Effizienz der DNA-Synthesechemie nicht 100 % beträgt.Dabei handelt es sich um einen zirkulären Prozess, der wiederholte chemische Reaktionen nutzt, während jede Base hinzugefügt wird, um DNA von 3′ bis 5′ herzustellen.Sie können in jedem Zyklus scheitern.Längere Primer, insbesondere solche mit mehr als 50 Basen, weisen einen großen Anteil verkürzter Sequenzen auf und müssen möglicherweise gereinigt werden.
Die Ausbeute an Primern wird durch die Effizienz der Synthesechemie und der Reinigungsmethode beeinflusst.Biopharmazeutische Unternehmen wie Cytology und Shengong verwenden alle eine minimale OD-Einheit, um die Gesamtproduktion von Oligonukleosiden sicherzustellen.Kundenspezifische Grundierungen werden in Trockenpulverform geliefert.Es ist am besten, die Primer in TE wieder aufzulösen, sodass die Endkonzentration 100 μM beträgt.TE ist besser als entionisiertes Wasser, da der pH-Wert des Wassers oft sauer ist und zur Hydrolyse von Oligonukleosiden führt.
Die Stabilität von Primern hängt von den Lagerbedingungen ab.Trockenes Pulver und gelöste Grundierungen sollten bei -20 °C gelagert werden.In TE gelöste Primer in Konzentrationen von mehr als 10 μM können bei -20 °C sechs Monate lang stabil gelagert werden, bei Raumtemperatur (15 °C bis 30 °C) jedoch nur weniger als eine Woche.Trockene Pulvergrundierungen können bei -20 °C mindestens 1 Jahr und bei Raumtemperatur (15 °C bis 30 °C) bis zu 2 Monate gelagert werden.
5. Enzyme und ihre Konzentrationen
Die derzeit verwendete Taq-DNA-Polymerase ist im Wesentlichen das gentechnische Enzym, das von coliformen Bakterien synthetisiert wird.Die zur Katalyse einer typischen PCR-Reaktion benötigte Enzymmenge beträgt etwa 2,5 U (bezogen auf das Gesamtreaktionsvolumen von 100 μl).Ist die Konzentration zu hoch, kann es zu einer unspezifischen Amplifikation kommen;Ist die Konzentration zu niedrig, verringert sich die Menge an synthetischem Produkt.
6. Qualität und Konzentration von dNTP
Die Qualität von dNTP hängt eng mit der Konzentration und der Effizienz der PCR-Amplifikation zusammen.Das dNTP-Pulver ist körnig und seine Variabilität verliert seine biologische Aktivität, wenn es unsachgemäß gelagert wird.Die dNTP-Lösung ist sauer und sollte in hoher Konzentration mit 1 M NaOH oder 1 M Tris.HCL-Pufferlösung verwendet werden, um den pH-Wert auf 7,0 bis 7,5 einzustellen. Kleine Menge Unterverpackung, Tiefkühllagerung bei -20 °C.Durch mehrfaches Einfrieren und Auftauen wird dNTP abgebaut.Bei der PCR-Reaktion sollte dNTP 50 bis 200 umol/L betragen.Insbesondere sollte darauf geachtet werden, dass die Konzentration der vier DNTPs gleich sein sollte (gleiche Molpräparation).Wenn sich die Konzentration einer von ihnen von der der anderen unterscheidet (höher oder niedriger), kommt es zu einer Nichtübereinstimmung.Eine zu niedrige Konzentration verringert die Ausbeute an PCR-Produkten.dNTP kann sich mit Mg2+ verbinden und die Konzentration an freiem Mg2+ verringern.
7. Template-Nukleinsäure (Zielgen).
Die Menge und der Reinigungsgrad der Template-Nukleinsäure sind einer der Schlüsselfaktoren für den Erfolg oder Misserfolg der PCR.Die traditionellen DNA-Reinigungsmethoden verwenden normalerweise SDS und Protease K, um Proben aufzuschließen und zu entsorgen.Die Hauptfunktionen von SDS sind: Auflösen von Lipiden und Proteinen auf der Zellmembran, wodurch die Zellmembran durch Auflösen von Membranproteinen zerstört wird, und Kernproteine in der Zelle dissoziieren, SDS kann sich auch mit Proteinen verbinden und ausfallen;Protease K kann Proteine, insbesondere an DNA gebundene Histone, hydrolysieren und verdauen und dann organische Lösungsmittel wie Phenol und Chloroform verwenden, um Proteine und andere Zellbestandteile zu extrahieren, und Ethanol oder Isopropylalkohol verwenden, um Nukleinsäure auszufällen.Die extrahierte Nukleinsäure kann als Vorlage für PCR-Reaktionen verwendet werden.Für allgemeine klinische Nachweisproben kann eine schnelle und einfache Methode verwendet werden, um Zellen aufzulösen, Krankheitserreger zu lysieren, Proteine aus Chromosomen zu verdauen und zu entfernen, um Zielgene freizusetzen, und direkt für die PCR-Amplifikation verwendet werden.Bei der RNA-Template-Extraktion wird normalerweise die Guanidinisothiocyanat- oder Protease-K-Methode verwendet, um zu verhindern, dass RNase RNA abbaut.
8.Mg2+-Konzentration
Mg2+ hat einen erheblichen Einfluss auf die Spezifität und Ausbeute der PCR-Amplifikation.Bei einer allgemeinen PCR-Reaktion beträgt die geeignete Konzentration von Mg2+ 1,5 bis 2,0 mmol/L, wenn die Konzentration verschiedener dNTP 200 umol/L beträgt.Ist die Mg2+-Konzentration zu hoch, nimmt die Reaktionsspezifität ab, es kommt zu unspezifischer Amplifikation, eine zu niedrige Konzentration verringert die Aktivität der Taq-DNA-Polymerase, was zu einer Reduzierung der Reaktionsprodukte führt.
Magnesiumionen beeinflussen mehrere Aspekte der PCR, wie z. B. die DNA-Polymeraseaktivität, die sich auf die Ausbeute auswirkt;Ein weiteres Beispiel ist das Primer-Annealing, das die Spezifität beeinflusst.dNTP und Templat binden an Magnesiumionen und reduzieren so die Menge an freien Magnesiumionen, die für die Enzymaktivität erforderlich ist.Die optimale Magnesiumionenkonzentration variiert für verschiedene Primerpaare und Templates, aber eine typische PCR-Startkonzentration mit 200 μM dNTP beträgt 1,5 mM (Hinweis: Für quantitative Echtzeit-PCR verwenden Sie 3 bis 5 mM Magnesiumionenlösung mit einer fluoreszierenden Sonde).Höhere Konzentrationen an freien Magnesiumionen erhöhen die Ausbeute, erhöhen aber auch die unspezifische Amplifikation und verringern die Wiedergabetreue.Um die optimale Konzentration zu bestimmen, wurden Magnesiumionentitrationen in Schritten von 0,5 mM von 1 mM bis 3 mM durchgeführt.Um die Abhängigkeit von der Magnesiumionenoptimierung zu verringern, kann Platinum Taq DNA-Polymerase verwendet werden.Die Platin-Taq-DNA-Polymerase ist in der Lage, ihre Funktion über einen größeren Bereich von Magnesiumionenkonzentrationen aufrechtzuerhalten als die Taq-DNA-Polymerase und erfordert daher weniger Optimierung.
9. PCR-fördernde Zusatzstoffe
Die Optimierung der Annealing-Temperatur, des Primerdesigns und der Magnesiumionenkonzentration reicht für eine hochspezifische Amplifikation der meisten Templates aus;Allerdings erfordern einige Vorlagen, darunter solche mit hohem GC-Gehalt, zusätzliche Maßnahmen.Additive, die die Schmelztemperatur der DNA beeinflussen, bieten eine weitere Möglichkeit, die Produktspezifität und -ausbeute zu verbessern.Für beste Ergebnisse ist eine vollständige Denaturierung der Vorlage erforderlich.
Darüber hinaus verhindert die Sekundärstruktur die Primerbindung und Enzymverlängerung.
PCR-Zusätze, einschließlich Formamid, DMSO, Glycerin, Betain und PCRx-Enhancer-Lösung, verbessern die Amplifikation.Ihr möglicher Mechanismus besteht darin, die Schmelztemperatur zu senken und so das Annealing von Primern und die Verlängerung der DNA-Polymerase durch den Sekundärstrukturbereich zu unterstützen.Die PCRx-Lösung bietet weitere Vorteile.Bei Verwendung mit Platinum Taq DNA-Polymerase und Platinum Pfx DNA-Polymerase ist eine minimale Magnesiumionenoptimierung erforderlich.Daher wird die Platintechnik mit dem Additiv kombiniert, um die Spezifität zu erhöhen und gleichzeitig die Abhängigkeit vom dritten Ansatz, der Magnesiumionenoptimierung, zu verringern.Um optimale Ergebnisse zu erzielen, sollte die Konzentration der Zusatzstoffe optimiert werden, insbesondere DMSO, Formamid und Glycerin, die die Taq-DNA-Polymerase hemmen.

10. Heißstart
Die Hot-Start-PCR ist neben einem guten Primer-Design eine der wichtigsten Methoden zur Verbesserung der PCR-Spezifität.Obwohl die optimale Verlängerungstemperatur der Taq-DNA-Polymerase 72 °C beträgt, bleibt die Polymerase bei Raumtemperatur aktiv.Somit entstehen unspezifische Produkte, wenn die Haltetemperatur während der Vorbereitung der PCR-Reaktion und zu Beginn des thermischen Zyklus niedriger als die Annealing-Temperatur ist.Sobald diese unspezifischen Produkte gebildet sind, werden sie wirksam verstärkt.Die Hot-Start-PCR ist besonders effektiv, wenn die für das Primer-Design verwendeten Stellen durch die Position genetischer Elemente begrenzt sind, wie z. B. ortsspezifische Mutationen, Expressionsklonierung oder Konstruktion und Manipulation genetischer Elemente, die für das DNA-Engineering verwendet werden.
Eine übliche Methode zur Begrenzung der Aktivität der Taq-DNA-Polymerase besteht darin, eine PCR-Reaktionslösung auf Eis vorzubereiten und sie in ein vorgeheiztes PCR-Gerät zu geben.Diese Methode ist einfach und kostengünstig, vervollständigt jedoch nicht die Aktivität des Enzyms und eliminiert daher die Amplifikation unspezifischer Produkte nicht vollständig.
Das thermische Priming verzögert die DNA-Synthese, indem es eine wesentliche Komponente hemmt, bis das PCR-Gerät die Denaturierungstemperatur erreicht.Die meisten manuellen thermischen Initiierungsmethoden, einschließlich der verzögerten Zugabe von Taq-DNA-Polymerase, sind umständlich, insbesondere für Anwendungen mit hohem Durchsatz.Andere thermische Grundierungsmethoden verwenden einen Wachsschutz, um eine wesentliche Komponente, einschließlich Magnesiumionen oder Enzyme, einzuschließen oder um reaktive Komponenten, wie z. B. Template und Puffer, physikalisch zu isolieren.Während des Wärmezyklus werden die verschiedenen Komponenten freigesetzt und beim Schmelzen des Wachses miteinander vermischt.Wie die manuelle Heißstartmethode ist die Wachsschutzmethode umständlich und anfällig für Verunreinigungen und eignet sich nicht für Anwendungen mit hohem Durchsatz.
Platin-DNA-Polymerase ist praktisch und effizient für die automatische Hot-Start-PCR.Die Platin-Taq-DNA-Polymerase besteht aus rekombinanter Taq-DNA-Polymerase in Kombination mit einem monoklonalen Antikörper gegen Taq-DNA-Polymerase.Die Antikörper werden durch PCR formuliert, um die Enzymaktivität bei längerer Temperaturhaltung zu hemmen.Taq-DNA-Polymerase wurde während der 94 °C-Isolierung des Denaturierungsschritts in die Reaktion freigesetzt, wodurch die volle Polymeraseaktivität wiederhergestellt wurde.Im Gegensatz zur chemisch modifizierten Taq-DNA-Polymerase zur thermischen Initiierung erfordert das Platin-Enzym keine längere Isolierung bei 94 °C (10 bis 15 Minuten), um die Polymerase zu aktivieren.Mit der PlatinumTaq-DNA-Polymerase wurden 90 % der Taq-DNA-Polymeraseaktivität nach 2 Minuten bei 94 °C wiederhergestellt.

11. Nest-PCR
Aufeinanderfolgende Amplifikationsrunden mit verschachtelten Primern können die Spezifität und Empfindlichkeit verbessern.Die erste Runde ist eine Standardverstärkung von 15 bis 20 Zyklen.Ein kleiner Teil des anfänglichen Amplifikationsprodukts wurde 100- bis 1000-fach verdünnt und der zweiten Amplifikationsrunde für 15 bis 20 Zyklen hinzugefügt.Alternativ kann die Größe des anfänglichen amplifizierten Produkts durch Gelreinigung bestimmt werden.In der zweiten Amplifikationsrunde wird ein verschachtelter Primer verwendet, der an die Zielsequenz innerhalb des ersten Primers binden kann.Die Verwendung der verschachtelten PCR verringert die Möglichkeit der Amplifikation mehrerer Zielstellen, da es nur wenige Zielsequenzen gibt, die zu beiden Primersätzen komplementär sind.Die gleiche Gesamtzahl an Zyklen (30 bis 40) mit den gleichen Primern amplifizierte die unspezifischen Stellen.Nested PCR erhöht die Empfindlichkeit begrenzter Zielsequenzen (z. B. seltene mrnas) und verbessert die Spezifität schwieriger PCRS (z. B. 5′ RACE).
12. Absteigende PCR
Die absteigende PCR verbessert die Spezifität, indem in den ersten PCR-Zyklen strenge Annealing-Bedingungen verwendet werden.Der Zyklus beginnt bei einer Glühtemperatur, die etwa 5 °C über der geschätzten Tm liegt. Anschließend wird jeder Zyklus um 1 °C bis 2 °C reduziert, bis die Glühtemperatur unter 5 °C Tm liegt.Nur die Zielvorlage mit der höchsten Homologie wird amplifiziert.Diese Produkte dehnen sich in nachfolgenden Zyklen weiter aus und verdrängen die verstärkten unspezifischen Produkte.Die absteigende PCR ist für Methoden nützlich, bei denen der Grad der Homologie zwischen Primer und Zieltemplate nicht bekannt ist, wie zum Beispiel AFLP-DNA-Fingerprinting.
Verwandte PCR-Kits
Der 2× PCR-HeldTMDas Mix-System hat eine höhere Toleranz gegenüber PCR-Inhibitoren als das gewöhnliche PCR-Mix-System und kann die PCR-Amplifikation verschiedener komplexer Templates problemlos bewältigen.Das einzigartige Reaktionssystem und der hocheffiziente Taq Hero sorgen dafür, dass die PCR-Reaktion eine höhere Amplifikationseffizienz, Spezifität und Empfindlichkeit aufweist.
Höhere Verstärkungseffizienz
Es verfügt über 5'→3'-DNA-Polymeraseaktivität und 5'→3'-Exonukleaseaktivität, ohne 3'→5'-Exonukleaseaktivität.
Real Time PCR Easyᵀᴹ-SYBR Green I Kit
Spezifisch – optimierter Puffer und Hot-Start-Taq-Enzym können unspezifische Amplifikation und Primer-Dimer-Bildung verhindern
Hohe Empfindlichkeit – kann geringe Kopien der Vorlage erkennen
Das Kit verwendet ein einzigartiges Foregene-Reverse-Transkriptionsreagenz und Foregene HotStar Taq DNA-Polymerase in Kombination mit einem einzigartigen Reaktionssystem, um die Amplifikationseffizienz und Spezifität der Reaktion effektiv zu verbessern.
Zeitpunkt der Veröffentlichung: 09.05.2023



