



1. Erstes Verständnis
In dieser Phase müssen wir einige Konzepte und Terminologie verstehen, um Fehler vor unseren Vorgesetzten zu vermeiden, wie zum Beispiel:
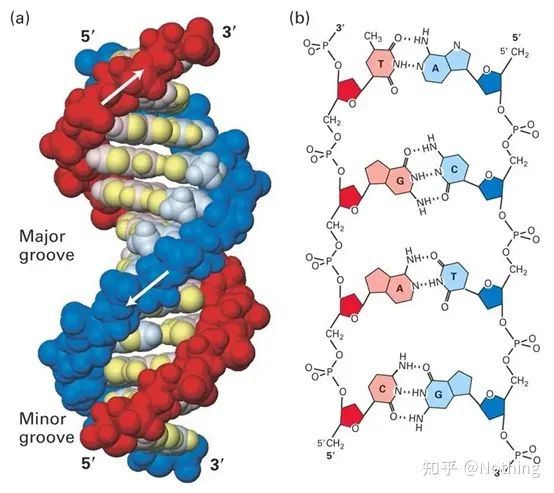
F: Was ist der Unterschied zwischen RT-PCR, qPCR, Echtzeit-PCR und Echtzeit-RT-PCR?
Antwort: RT-PCR ist eine Reverse-Transkriptions-PCR(Reverse Transkriptions-PCR, RT-PCR), eine weit verbreitete Variante der Polymerase-Kettenreaktion (PCR).Bei der RT-PCR wird ein RNA-Strang revers in komplementäre DNA transkribiert, die dann als Matrize für die DNA-Amplifikation durch PCR verwendet wird.
Echtzeit-PCR und qPCR(Quantitative Realtime-PCR) sind dasselbe, beide sind quantitative Echtzeit-PCR, was bedeutet, dass jeder PCR-Zyklus über Echtzeitdatenaufzeichnungen verfügt, sodass die Anzahl der Startvorlagen für eine präzise Analyse angepasst werden kann.
Obwohl sowohl die Echtzeit-PCR (quantitative Echtzeit-Fluoreszenz-PCR) als auch die Reverse-Transkriptions-PCR (Reverse-Transkriptions-PCR) als RT-PCR abgekürzt zu werden scheinen, lautet die internationale Konvention: RT-PCR bezieht sich speziell auf die umgekehrte TranskriptionPCR, Echtzeit-PCR wird im Allgemeinen als qPCR (quantitative Echtzeit-PCR) abgekürzt..
Und Echtzeit-RT-PCR (RT-qPCR). Hierbei handelt es sich um die Reverse-Transkriptions-PCR in Kombination mit der quantitativen Fluoreszenztechnologie: Erhalten Sie zunächst cDNA (RT) aus der reversen RNA-Transkription und verwenden Sie dann Echtzeit-PCR für die quantitative Analyse (qPCR).Die meisten Labore führen RT-qPCR durch, also Untersuchungen zur Herunterregulierung der RNA-Expression. Die qPCR, über die im Labor alle reden, bezieht sich also eigentlich auf RT-qPCR, aber vergessen Sie nicht, dass es immer noch viele DNA-Tests in klinischen Anwendungen gibt.Quantitative Analyse, z. B. Nachweis des Hepatitis-B-Virus-HBV.
Frage: Warum sollte das amplifizierte Fragment nach dem Auslesen vieler quantitativer Fluoreszenz-PCR im Bereich von 80–300 bp kontrolliert werden?
Antworten: Die Länge jeder Gensequenz ist unterschiedlich, einige sind mehrere kb, andere sind Hunderte von bp, aber wir müssen beim Entwerfen von Primern nur eine Produktlänge von 80-300 bp verlangen, zu kurz oder zu lang sind nicht für den fluoreszierenden quantitativen PCR-Nachweis geeignet.Das Produktfragment ist zu kurz, um vom Primer-Dimer unterschieden zu werden.Die Länge des Primer-Dimers beträgt etwa 30–40 bp, und es ist schwierig zu unterscheiden, ob es sich um ein Primer-Dimer oder ein Produkt handelt, wenn es weniger als 80 bp beträgt.Wenn das Produktfragment zu lang ist und 300 bp überschreitet, führt dies leicht zu einer geringen Amplifikationseffizienz und kann die Menge des Gens nicht effektiv erfassen.
Wenn Sie beispielsweise zählen, wie viele Personen sich in einem Klassenzimmer befinden, müssen Sie nur zählen, wie viele Münder es gibt.Das Gleiche gilt, wenn Sie Gene erkennen. Sie müssen nur eine bestimmte Sequenz eines Gens erkennen, um sie darzustellen. Die gesamte Sequenz reicht aus.Wenn Sie Menschen zählen möchten, müssen Sie sowohl Münder als auch Nasen, Ohren und Brillen zählen, und es ist leicht, Fehler zu machen.
Darüber hinaus gibt es in der biologischen Forschung viele Forschungsfälle von Punkt zu Bereich, da die Gensequenz jeder Art sehr lang ist und es unnötig und unmöglich ist, alle Fragmente zu messen, wie z. B. die bakterielle 16S-Sequenzierung, bei der die konservative Reihenfolge von Bakterien durchgeführt wird. Tests, um auf die Anzahl einer bestimmten Bakterienpopulation zu schließen.
F: Was ist die optimale Länge für das qPCR-Primerdesign?
Antworten: Im Allgemeinen beträgt die Primerlänge etwa 20-24 bp, was besser ist.Natürlich müssen wir beim Entwerfen des Primers auf den TM-Wert des Primers achten, da dieser mit der optimalen Glühtemperatur zusammenhängt.Nach vielen Experimenten wurde bewiesen, dass 60°C ein besserer TM-Wert ist.Wenn die Annealing-Temperatur zu niedrig ist, kommt es leicht zu einer unspezifischen Amplifikation.Wenn die Glühtemperatur zu hoch ist, ist die Amplifikationseffizienz relativ niedrig, der Höhepunkt der Amplifikationskurve beginnt später und der CT-Wert wird verzögert.
F: Wie unterscheidet sich die Färbemethode von der Sondenmethode?
Antwort: FärbemethodeEinige Fluoreszenzfarbstoffe wie SYBR Green Ⅰ, PicoGreen, BEBO usw. emittieren selbst kein Licht, sondern emittieren Fluoreszenz, nachdem sie an die kleine Furche doppelsträngiger DNA gebunden sind.Daher kann das Gerät zu Beginn der PCR-Reaktion das Fluoreszenzsignal nicht erkennen.Wenn die Reaktion das Annealing-Extension-Stadium erreicht, wird der Doppelstrang geöffnet und unter der Wirkung der DNA-Polymerase ein neuer Strang synthetisiert, und das fluoreszierende Molekül bindet an die kleine Furche der dsDNA.Mit zunehmender Anzahl der PCR-Zyklen werden immer mehr Farbstoffe mit doppelsträngiger DNA kombiniert und auch das Fluoreszenzsignal wird kontinuierlich verstärkt.Die Färbemethode wird hauptsächlich in der wissenschaftlichen Forschung eingesetzt.
PS: Seien Sie bei der Durchführung des Experiments vorsichtig. Der Farbstoff muss mit menschlicher DNA kombiniert werden. Achten Sie darauf, ihn in eine fluoreszierende Person umzuwandeln.
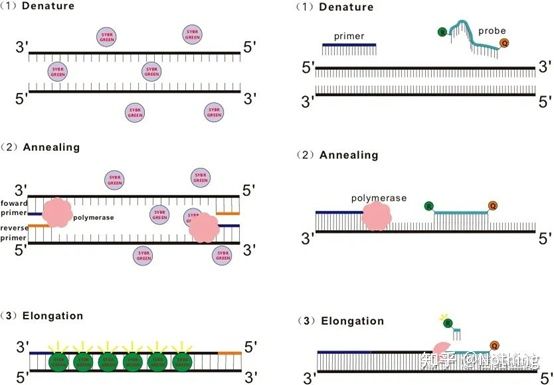
Farbstoffmethode (links) Sondenmethode (rechts)
PS: Seien Sie bei der Durchführung des Experiments vorsichtig. Der Farbstoff muss mit menschlicher DNA kombiniert werden. Achten Sie darauf, ihn in eine fluoreszierende Person umzuwandeln.
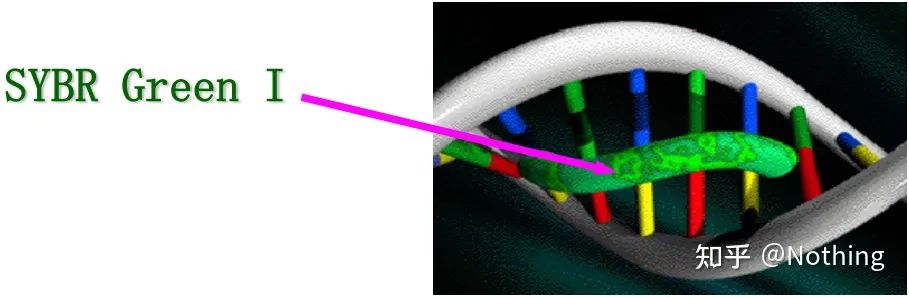
SYBR Green Ⅰ bindet an die kleine Furche der DNA
SondenmethodeDie Taqman-Sonde ist die am häufigsten verwendete Hydrolysesonde.Am 5′-Ende der Sonde befindet sich eine fluoreszierende Gruppe, normalerweise FAM, und die Sonde selbst ist eine zum Zielgen komplementäre Sequenz.Am 3′-Ende befindet sich eine Fluoreszenzlöschgruppe.Nach dem Prinzip des Fluoreszenzresonanzenergietransfers (Förster-Resonanzenergietransfer, FRET) kann die Anregung des Donormoleküls die Fluoreszenz des Akzeptormoleküls induzieren, wenn die Reporter-Fluoreszenzgruppe (Donor-Fluoreszenzmolekül) und die löschende Fluoreszenzgruppe (Akzeptor-Fluoreszenzmolekül) angeregt werden. Wenn sich die Spektren überlappen und der Abstand sehr gering ist (7–10 nm).Daher emittiert die Reporter-Fluoreszenzgruppe zu Beginn der PCR-Reaktion keine Fluoreszenz, wenn die Sonde im System frei und intakt ist.Beim Annealing binden Primer und Sonde an die Matrize.Während der Verlängerungsphase synthetisiert die Polymerase kontinuierlich neue Ketten.DNA-Polymerase hat eine 5′-3′-Exonukleaseaktivität.Beim Erreichen der Sonde hydrolysiert die DNA-Polymerase die Sonde von der Matrize, trennt die Reporter-Fluoreszenzgruppe von der Quencher-Fluoreszenzgruppe und setzt das Fluoreszenzsignal frei.Da zwischen der Sonde und der Vorlage eine Eins-zu-eins-Beziehung besteht, ist die Sondenmethode der Farbstoffmethode hinsichtlich der Genauigkeit und Empfindlichkeit des Tests überlegen.Die Sondenmethode wird hauptsächlich in der Diagnose eingesetzt.
F: Was ist absolute Quantifizierung?Was ist relative Quantifizierung?
Antworten: Die absolute Quantifizierung bezieht sich auf die Berechnung der anfänglichen Kopienzahl der durch qPCR zu testenden Probe, z. B. wie viele HBV-Viren in 1 ml Blut enthalten sind.Das durch relative Quantifizierung erhaltene Ergebnis ist die Änderung der Menge des Zielgens in einer bestimmten Probe im Vergleich zu einer anderen Referenzprobe, und die Genexpression wird hoch- oder herunterreguliert.
F: Werden sich die Menge der RNA-Extraktion, die Effizienz der reversen Transkription und die Amplifikationseffizienz auf die experimentellen Ergebnisse auswirken?
F: Beeinflussen Probenlagerung, Extraktionsreagenzien, Reverse-Transkriptions-Reagenzien und lichtdurchlässige Verbrauchsmaterialien die Versuchsergebnisse?
F: Mit welcher Methode können die experimentellen Daten korrigiert werden?
Bezüglich dieser Probleme werden wir sie in den Abschnitten „Erweitert“ und „Erweitert“ weiter unten ausführlich beschreiben.
2. Fortgeschrittene Kenntnisse
Im Hinblick auf die quantitative Echtzeit-Fluoreszenz-PCR müssen wir uns darüber im Klaren sein, dass jedes Jahr Tausende von wissenschaftlichen Forschungsarbeiten veröffentlicht werden, von denen die quantitative Fluoreszenz-PCR-Technologie keine kleine Zahl darstellt.
Wenn es keinen gemeinsamen Standard zur Messung des quantitativen Fluoreszenz-PCR-Experiments gibt, können die Ergebnisse stark variieren.Für dasselbe Gen derselben Art und mit derselben Verarbeitungsmethode variieren die Nachweisergebnisse ebenfalls erheblich, und es wird für Nachzügler schwierig sein, dieselben Ergebnisse zu wiederholen.Niemand weiß, was richtig und was falsch ist.
Bedeutet das, dass die quantitative Fluoreszenz-PCR eine Betrugstechnologie oder eine unzuverlässige Technologie ist?Nein, das liegt daran, dass die quantitative Fluoreszenz-PCR empfindlicher und genauer ist und eine kleine falsche Bedienung zu völlig gegenteiligen Ergebnissen führt.Ein kleiner Verlust ist tausend Meilen entfernt.Der Autor des Artikels wird möglicherweise wiederholt von den Rezensenten gefoltert.Gleichzeitig fällt es den Gutachtern der Zeitschrift auch schwer, aus unterschiedlichen experimentellen Ergebnissen auszuwählen.
Alles in allem deutet dies auf einen mangelnden Konsens bei Echtzeit-PCR-Experimenten hin.Zu diesem Zweck begannen hochrangige Wissenschaftler der Branche, Standards zu formulieren.Von den Mitwirkenden wird verlangt, dass sie in dem Artikel einige notwendige experimentelle und Datenverarbeitungsdetails (einschließlich notwendiger Daten) angeben, um diese Standards zu erfüllen.
Gutachter können die Qualität des Experiments beurteilen, indem sie diese Details lesen;Zukünftige Leser können dies auch nutzen, um das Experiment zu wiederholen oder zu verbessern.Dann sind die so gewonnenen experimentellen Ergebnisse aussagekräftig, hochwertig und nutzbar.
MIBBI (Mindestinformationen für biologische und biomedizinische Untersuchungen –http://www.mibbi.org) entstand.MIBBI ist ein Projekt, das Standards für Experimente bereitstellt.Es wird in der Natur veröffentlicht.Dieses Projekt zielt auf verschiedene biologische Experimente ab, darunter Zellbiologie, Microarray, qPCR, die wir jetzt besprechen werden, usw., und berücksichtigt bei der Einreichung von Manuskripten jede Art von Experiment.Diese Informationen sollten jederzeit bereitgestellt werden.
Im MIBBI-Projekt gibt es zwei Artikel, die sich auf die quantitative Fluoreszenz-PCR beziehen, nämlich:
·RDML (Real-Time PCR Data Markup Language) – eine strukturierte Sprache und ein Berichtsleitfaden für quantitative Echtzeit-PCR-Daten;
·MIQE (Mindestinformationen für die Veröffentlichung quantitativer Echtzeit-PCR-Experimente) – Mindestinformationen für die Veröffentlichung von Artikeln über quantitative Echtzeit-PCR-Experimente.
Lassen Sie uns zunächst über RDML sprechen, die Terminologiespezifikation.
Wenn es keine einheitliche Definition für alles gibt, ist eine Fortsetzung der Diskussion nicht möglich, weshalb die Erklärung von Begriffen in der Prüfung so wichtig ist.
Die im Fluoreszenz-quantitativen PCR-Experiment verwendete Terminologie umfasst den folgenden Inhalt.QIAGEN hat für uns die beste Zusammenfassung erstellt.Die folgenden sind alle trockenWaren .
Verstärkungskurve
Die Amplifikationskurve bezieht sich auf die Kurve, die während des PCR-Prozesses erstellt wurde, mit der Zyklusnummer als Abszisse und der Echtzeit-Fluoreszenzintensität während der Reaktion als Ordinate.
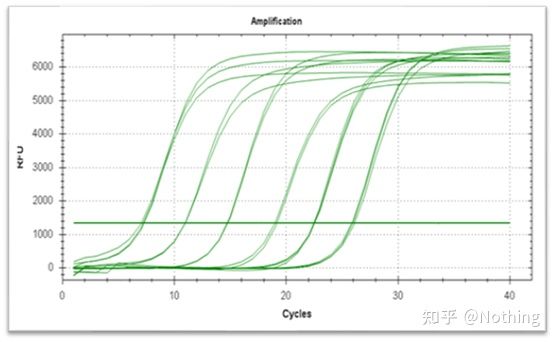
Eine hervorragende Verstärkungskurve sollte die folgenden Eigenschaften aufweisen: Die Grundlinie ist flach oder leicht gesunken und es gibt keinen offensichtlichen Aufwärtstrend;Der Wendepunkt der Kurve ist deutlich zu erkennen und die Steigung der Exponentialphase ist proportional zur Verstärkungseffizienz.Je größer die Steigung, desto höher die Verstärkungseffizienz;die Gesamtverstärkungskurve Die Parallelität ist gut, was darauf hinweist, dass die Verstärkungseffizienz jeder Röhre ähnlich ist;Die exponentielle Phase der Amplifikationskurve von Proben mit niedriger Konzentration ist offensichtlich.
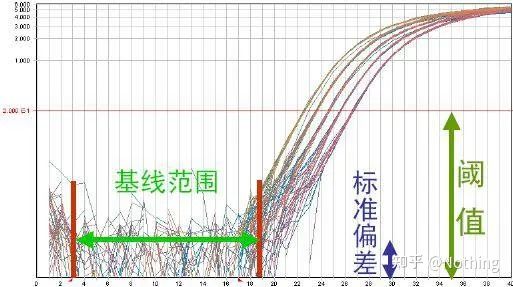
Grundlinie (Grundlinie)
Die Basislinie ist der Geräuschpegel des frühen Zyklus, üblicherweise zwischen dem 3. und 15. Zyklus gemessen, da der durch das Amplifikationsprodukt verursachte Fluoreszenzwertanstieg in diesem Zeitraum nicht nachgewiesen werden kann.Die Anzahl der zur Berechnung der Basislinie verwendeten Zyklen kann variiert werden und muss möglicherweise reduziert werden, wenn hohe Template-Mengen verwendet werden oder wenn das Expressionsniveau des Zielgens hoch ist.
Zum Festlegen der Grundlinie müssen die Fluoreszenzdaten aus der Linearitätsverstärkungskurve betrachtet werden.Die Basislinie wird so eingestellt, dass das Wachstum der Amplifikationskurve mit einer Zykluszahl beginnt, die größer ist als die obere Zykluszahl der Basislinie.Basislinien müssen für jede Zielsequenz individuell festgelegt werden.Die in den frühen Zyklen erfassten durchschnittlichen Fluoreszenzwerte müssen von den in den amplifizierten Produkten erhaltenen Fluoreszenzwerten abgezogen werden.Die neuesten Versionen verschiedener Real-Time-PCR-Software ermöglichen die automatische Optimierung der Basiseinstellungen für einzelne Proben.
Während der ersten paar Zyklen der PCR-Amplifikationsreaktion ändert sich das Fluoreszenzsignal nicht wesentlich.Die Annäherung an eine gerade Linie wird als Grundlinie bezeichnet, aber wenn wir uns die ersten paar Zyklen genau ansehen, sehen wir, dass sich innerhalb der Grundlinie das abspielt, was im Bild unten zu sehen ist.
Hintergrund Hintergrund bezieht sich auf
der unspezifische Fluoreszenzwert in der Reaktion.Zum Beispiel: ineffiziente Fluoreszenzlöschung;oder eine große Anzahl doppelsträngiger DNA-Matrizen aufgrund der Verwendung von SYBR Green.Die Hintergrundkomponenten des Signals werden durch den Echtzeit-PCR-Softwarealgorithmus mathematisch entfernt.
Reportersignal
Das Reportersignal bezieht sich auf das Fluoreszenzsignal, das von SYBR Green oder fluoreszenzmarkierten sequenzspezifischen Sonden während der Echtzeit-PCR erzeugt wird.
Normalisiertes Reportersignal (RN)
RN bezieht sich auf die Fluoreszenzintensität des Reporterfarbstoffs dividiert durch die Fluoreszenzintensität des passiven Referenzfarbstoffs, gemessen bei jedem Zyklus.
Passiver Referenzfarbstoff
Bei manchen Real-Time-PCRsDer Fluoreszenzfarbstoff ROX wird als interne Referenz zur Normalisierung des Fluoreszenzsignals verwendet.Es korrigiert Abweichungen aufgrund ungenauer Pipettierung, Well-Position und Fluoreszenzschwankungen auf Well-für-Well-Basis.

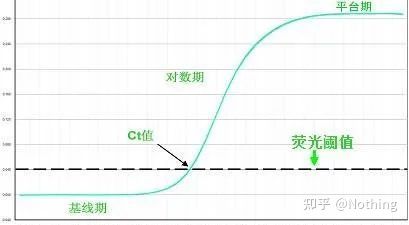
Die Fluoreszenzschwelle (Threshold)
wurde über den Hintergrundwert und deutlich unter den Plateauwert der Amplifikationskurve eingestellt.Er muss im linearen Bereich der Amplifikationskurve liegen, was den logarithmisch linearen Bereich der PCR-Detektion darstellt.In der Ansicht der logarithmischen Amplifikationskurve sollten Schwellenwerte festgelegt werden, damit die logarithmisch-lineare Phase der PCR leicht erkennbar ist.Wenn es in der Echtzeit-PCR mehrere Zielgene gibt, muss der Schwellenwert für jedes Ziel festgelegt werden.Im Allgemeinen wird das Fluoreszenzsignal der ersten 15 Zyklen der PCR-Reaktion als Fluoreszenz-Hintergrundsignal verwendet, und der Fluoreszenzschwellenwert beträgt das Zehnfache der Standardabweichung des Fluoreszenzsignals der ersten 3 bis 15 Zyklen der PCR, und der Fluoreszenzschwellenwert wird in der exponentiellen Phase der PCR-Amplifikation festgelegt.Im Allgemeinen wird vor der Verwendung jedes Instruments seine Fluoreszenzschwelle eingestellt.
Zyklusschwelle (CT) oder Kreuzungspunkt (CP)
Der Zyklus, bei dem die Amplifikationskurve den Schwellenwert überschreitet (d. h. der Punkt, an dem die Fluoreszenzerkennung deutlich zunimmt).CT kann ein Bruchteil sein und die Menge der Ausgangsvorlage kann berechnet werden.Der CT-Wert stellt die Anzahl der Zyklen dar, die auftreten, wenn das Fluoreszenzsignal in jedem PCR-Reaktionsröhrchen den eingestellten Schwellenwert erreicht.Es besteht eine lineare Beziehung zwischen dem CT-Wert jeder Vorlage und dem Logarithmus der anfänglichen Kopienzahl der VorlageJe höher die anfängliche Kopienzahl, desto kleiner der CT-Wert und umgekehrt.Eine Standardkurve kann unter Verwendung eines Standards mit bekannter Anfangskopienzahl erstellt werden, wobei die Abszisse den CT-Wert und die Ordinate den Logarithmus der Anfangskopienzahl darstellt.Solange der CT-Wert der unbekannten Probe erhalten wird, kann daher die anfängliche Kopienzahl der Probe aus der Standardkurve berechnet werden.
ΔCT-Wert
ΔCT-Wert beschreibtdie Differenz zwischen dem Zielgen und dem entsprechenden endogenen Referenzgen-CT-WertB. ein Housekeeping-Gen, und wird verwendet, um die Menge der verwendeten Vorlage zu normalisieren:
⇒ΔCT = CT (Zielgen) – CT (endogenes Referenzgen)
ΔΔCT-Wert
Der ΔΔCT-Wert beschreibt die Differenz zwischen dem mittleren ΔΔCT-Wert einer interessierenden Probe (z. B. stimulierten Zellen) und dem mittleren ΔΔCT-Wert einer Referenzprobe (z. B. nicht stimulierte Zellen).Die Referenzprobe wird auch als Kalibrierprobe bezeichnet und alle anderen Proben werden zur relativen Quantifizierung darauf normiert:
⇒ΔΔCT = durchschnittlicher ΔCT (interessierende Probe) – durchschnittlicher ΔCT (Referenzprobe)
Endogene Referenzgene (endogene Referenzgene)
Die Expressionsniveaus endogener Referenzgene, wie z. B. Housekeeping-Gene (Housekeeping-Gene), unterscheiden sich zwischen den Proben nicht.Durch den Vergleich der CT-Werte des Referenzgens mit dem Zielgen kann das Expressionsniveau des Zielgens auf die Menge der eingegebenen RNA oder cDNA normalisiert werden (siehe Abschnitt über ΔCT-Werte oben).
Interne Referenzgene korrigierenmöglicher RNA-Abbau oder das Vorhandensein von Enzyminhibitoren in RNA-Proben sowie Variationen im RNA-Gehalt, der Effizienz der reversen Transkription, der Nukleinsäuregewinnung und der Probenhandhabung.Um die optimalen Referenzgene auszuwählen, haben wir den Algorithmus geändert, um die Auswahl der optimalen Referenz abhängig von der experimentellen Einstellung zu ermöglichen.
Interne Kontrolle
Eine Kontrollsequenz, die in derselben Reaktion wie die Zielsequenz amplifiziert und mit einer anderen Sonde sondiert wird (z. B. bei der Durchführung einer Duplex-PCR).Interne Kontrollen werden häufig verwendet, um fehlgeschlagene Amplifikationen auszuschließen, beispielsweise wenn die Zielsequenz nicht erkannt wird.
Kalibrierungsprobe
Eine Referenzprobe (z. B. gereinigte RNA aus einer Zelllinie oder einem Gewebe), die bei der relativen Quantifizierung zum Vergleich aller anderen Proben verwendet wird, um das relative Expressionsniveau eines Gens zu bestimmen.Bei der Kalibrierungsprobe kann es sich um eine beliebige Probe handeln, in der Regel handelt es sich jedoch um eine Kontrolle (z. B. eine unbehandelte Probe oder eine Probe vom Zeitpunkt Null des Experiments).
Positivkontrollen
Verwenden Sie Kontrollreaktionen mitbekannte Mengen an Vorlage.Positivkontrollen werden häufig verwendet, um zu überprüfen, ob ein Primer-Set oder Primer-Sonden-Set ordnungsgemäß funktioniert und die Reaktion korrekt eingerichtet ist.
Keine Vorlagenkontrolle (NTC)
Eine Kontrollreaktion, die alle notwendigen Komponenten der Amplifikationsreaktion enthält, mit Ausnahme der Matrize, die normalerweise durch Wasser ersetzt wird.Der Einsatz von NTC kann die durch Reagenzverunreinigungen oder Fremd-DNA verursachte Kontamination aufdecken und so die Authentizität und Zuverlässigkeit der Nachweisdaten gewährleisten.Eine Verstärkung der NTC-Kontrolle weist auf eine Kontamination hin.
Keine RT-Kontrolle (NRT)
Der RNA-Extraktionsprozess kann restliche genomische DNA enthalten, die äußerst schädlich ist und die Datenqualität beeinträchtigt und den natürlichen Feind von qPCR darstellt. Daher muss bei der Gestaltung von Experimenten darauf geachtet werden, dass nur die RNA-Erkennung verstärkt wird.Es gibt zwei Möglichkeiten: Die eine besteht darin, Primer über Introns hinweg zu entwerfen, die andere darin, die DNA vollständig zu entfernen. Welche Methode ist besser und wird später besprochen.Die NTR-Steuerung ist ein Zauberspiegel zur Erkennung von DNA-Verschmutzungen.Wenn es eine Verstärkung gibt, bedeutet das, dass eine Verschmutzung vorliegt.
Standards
Standards sind Proben bekannter Konzentration oder Kopienzahl, die zur Erstellung einer Standardkurve verwendet werden.Um die Stabilität des Standards zu gewährleisten, wird das Genfragment üblicherweise in das Plasmid kloniert und als Standard verwendet.
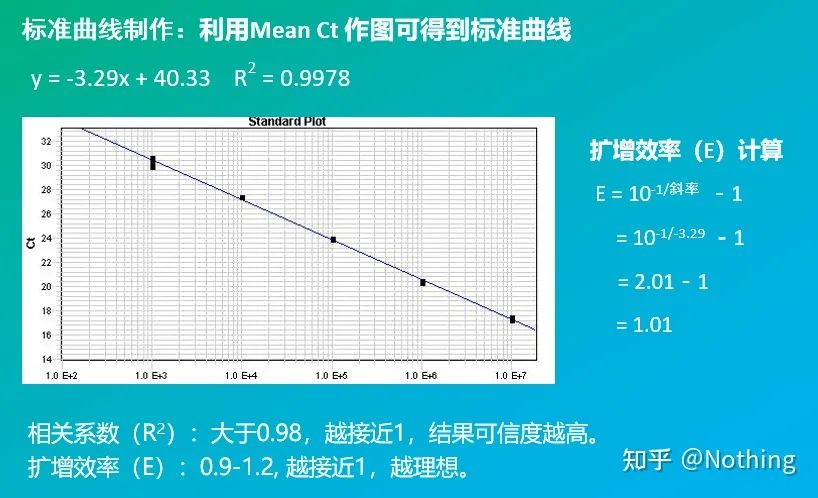
Die Standardkurve
wird normalerweise entsprechend dem Verdopplungsverhältnis in mindestens 5 Konzentrationsgradienten mit dem Standardprodukt verdünnt, und 5 Punkte werden in den Koordinaten von CT-Wert und Kopienzahl gezeichnet und die Punkte werden zu einer Linie verbunden, um eine Standardkurve zu erzeugen.Für jede Standardkurve muss ihre Gültigkeit überprüft werden.Der Steigungswert liegt zwischen –3,3 und –3,8 und jede Konzentration wird dreifach durchgeführt.Punkte, die sich deutlich von anderen Punkten unterscheiden, sollten verworfen werden.Der CT-Wert der zu testenden Probe wird in die Standardkurve eingebracht und das Expressionsniveau der zu testenden Probe kann berechnet werden.
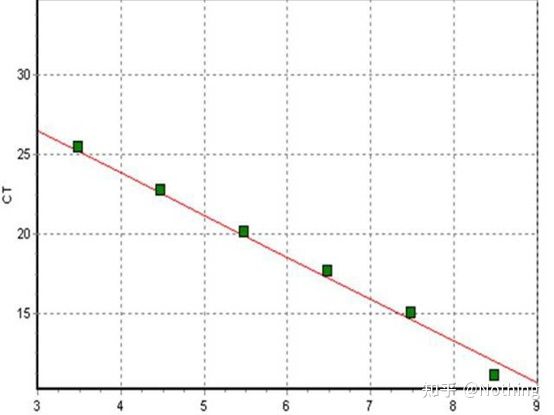
Der CT-Wert der zu untersuchenden Probe wird in die Standardkurve eingebracht und die anfängliche Kopienzahl der zu untersuchenden Probe kann berechnet werden.
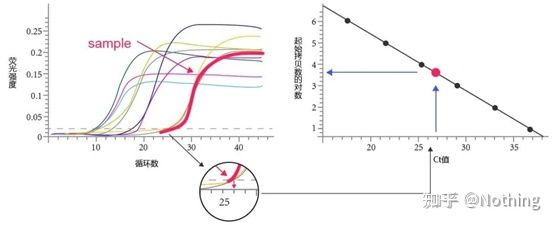
Effizienz und Steigung
Die Steigung der Standardkurve repräsentiert die Effizienz der Echtzeit-PCR.
·Eine Steigung von -3,322 zeigt an, dass die PCR-Amplifikationseffizienz 1 oder 100 % effizient ist und sich die Menge an PCR-Produkt bei jedem Zyklus verdoppelt.
·Eine Steigung von weniger als –3,322 (z. B. –3,8) weist auf eine PCR-Effizienz hin
·Eine Steigung von mehr als –3,322 (z. B. –3,0) zeigt an, dass die PCR-Effizienz größer als 100 % zu sein scheint. Das ist merkwürdig: Wie könnte ein PCR-Zyklus mehr als das Doppelte des amplifizierten Produkts erzeugen?Diese Situation tritt in der nichtlinearen Phase der PCR-Reaktion auf, das heißt, es findet eine große Menge unspezifischer Amplifikation statt.
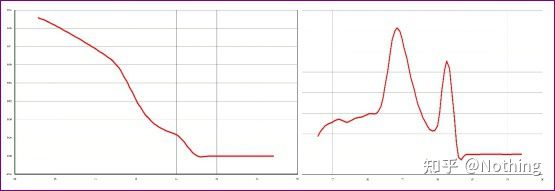
Schmelzkurve
Nachdem die qPCR-Amplifikation abgeschlossen ist, wird das PCR-Produkt erhitzt.Mit steigender Temperatur schmilzt das doppelsträngige Amplifikationsprodukt allmählich, was zu einer Abnahme der Fluoreszenzintensität führt.Wenn eine bestimmte Temperatur (Tm) erreicht wird, schmilzt eine große Anzahl von Produkten.Die Fluoreszenz nimmt stark ab.Verschiedene PCR-Produkte haben unterschiedliche Tm-Werte und unterschiedliche Schmelztemperaturen, sodass die Spezifität der PCR identifiziert werden kann.
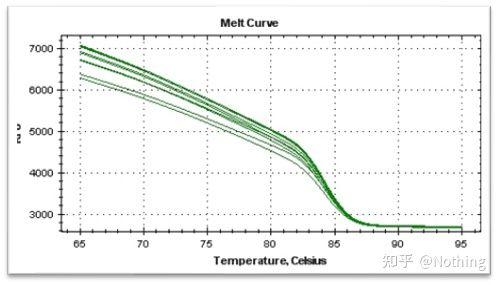
Schmelzkurve (Ableitungskurve)
Die Schmelzkurve wird abgeleitet, um eine Peakkarte zu erstellen, die die Situation von PCR-Produktfragmenten intuitiver darstellen kann.Da die Schmelztemperatur der Tm-Wert des DNA-Fragments ist, können einige Parameter beurteilt werden, die den Tm-Wert des DNA-Fragments beeinflussen, wie z. B. Fragmentgröße, GC-Gehalt usw. Im Allgemeinen gilt gemäß unseren Primer-Designprinzipien:Die Länge des amplifizierten Produkts liegt im Bereich von 80–300 bp, daher sollte die Schmelztemperatur zwischen 80 °C und 90 °C liegen.
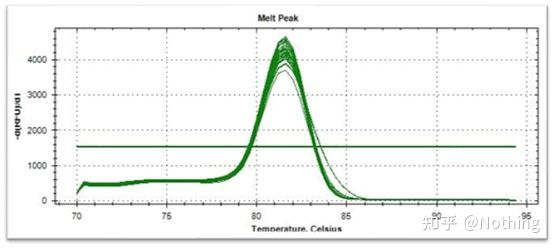
Interpretation der Schmelzkurve: Wenn der einzige Hauptpeak zwischen 80 °C und 90 °C erscheint, bedeutet dies, dass die quantitative Fluoreszenz-PCR perfekt ist;Wenn der Hauptpeak zwischen 80 °C und 90 °C auftritt und verschiedene Peaks unter 80 °C auftreten, wird grundsätzlich das Primer-Dimer in Betracht gezogen.Sie können versuchen, die Glühtemperatur zu erhöhen, um das Problem zu lösen.Wenn der Hauptpeak zwischen 80 °C und 90 °C erscheint und der sonstige Peak erneut erscheint, wenn die Temperatur steigt, wird grundsätzlich davon ausgegangen, dass eine DNA-Kontamination vorliegt und die DNA in der Anfangsphase des Experiments entfernt werden muss.
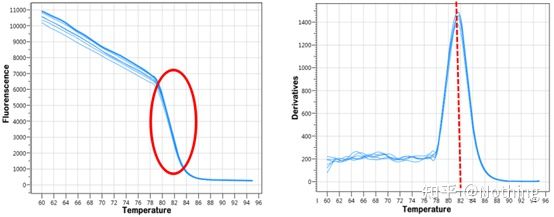
Natürlich gibt es immer noch einige ungewöhnliche Situationen, die im Folgenden einzeln aufgeschlüsselt werden.
3. Fortgeschrittene Kenntnisse
Um qPCR durchzuführen, muss ich MIQE sagen,Mindestinformationenzur Veröffentlichung vonQuantitativEchtzeit-PCRExperimente – die Mindestinformationen für die Veröffentlichung von Artikeln über quantitative Echtzeit-PCRExperimente.Um das Verständnis für alle zu vereinfachen, werden wir die wichtigsten Inhalte vereinfachen.
Sie können den Originaltext von MIQE im Internet durchsuchen. Das Wichtigste ist, dass darin festgelegt istDatencheckliste, die bei der Veröffentlichung eines Artikels bereitgestellt werden muss .
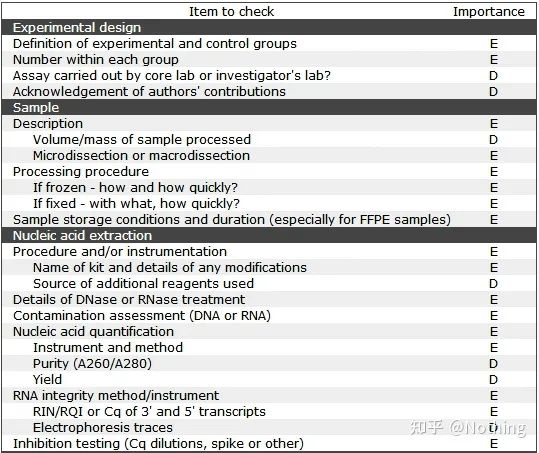
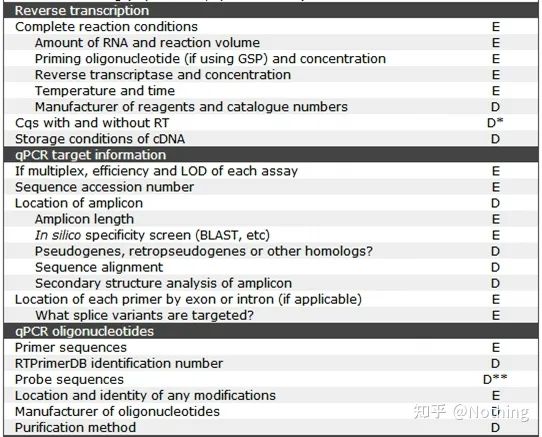
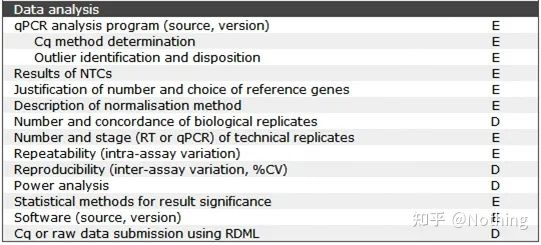
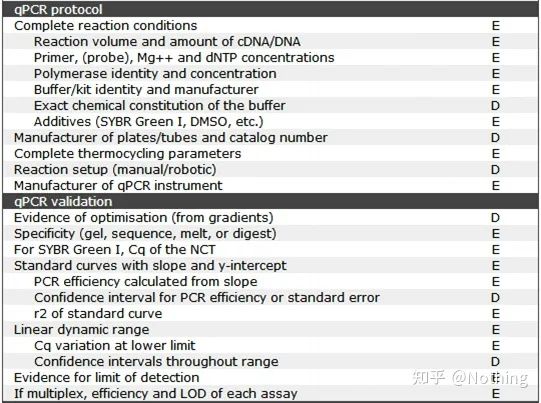
Gutachter können die Qualität des Experiments beurteilen, indem sie diese Details lesen;Zukünftige Leser können dies auch nutzen, um das Experiment zu wiederholen oder zu verbessern.
Es ist erwähnenswert, dass in dieser Liste die Wichtigkeit jeder Liste mit E bzw. D gekennzeichnet ist.Was bedeutet das?E: wesentliche Informationen (müssen eingereicht werden);D: wünschenswerte Informationen (so viel wie möglich bereitstellen).
MIQE (1) – Experimentelles Design
Viele Drecksäcke, die ihre Verteidigung nach dem Abschluss ihres Studiums abgeschlossen haben, wissen nicht, wie sie selbstständig ein Experiment entwerfen, ihre Notizbücher öffnen und tun können, was der Lehrer ihnen sagt.Infolgedessen war das experimentelle Design nicht streng, und die Redaktion des Magazins sagte, sie wolle dieses und jenes Bild erfinden, also taten sie es benommen.So werden die Drecksäcke gemacht!

Näher zu Hause besteht das erste Prinzip des Experiments darin, zu bestimmendie Strenge der experimentellen Logik.Das Wichtigste am experimentellen Design ist die Festlegung der Zielprobe, der Referenzprobe (Kontrolle) und der Anzahl der Wiederholungen, damit die experimentellen Daten referenziert, vergleichbar und überzeugend sind.
Die Zielprobebezieht sich auf die Probe, bei der wir nach einer bestimmten Behandlung das Zielgen nachweisen müssen.Das Referenzmusterist die Probe ohne jegliche Behandlung, die in der Biologie oft als Wildtyp bezeichnet wird.
Experimentelle Replikatesind sehr wichtig.Im Allgemeinen muss die Anzahl der überzeugenden Replikate mehr als drei betragen.Es muss unterschieden werden, was biologische Replikation und was technische Replikation ist.
Biologische Replikate: Das gleiche Verifizierungsexperiment wurde mit unterschiedlichen Materialien durchgeführt (Zeit, Anlagen, Chargen, Reaktionsplatten).

Biologische Vervielfältigung
Nehmen wir als Beispiel die Pestizidbehandlung von Pfeffer.Wir wollen Pestizide auf die drei Pflanzen von ABC sprühen, dann sind die drei Pflanzen von ABC drei biologische Replikate und es handelt sich um dasselbe Verifizierungsexperiment, das mit unterschiedlichen Materialien durchgeführt wurde.Aber als Experiment ist auf jeden Fall eine Kontrolle erforderlich, sodass wir einen der Zweige von Pflanze A besprühen können, um eine Versuchsgruppe von Pflanze A zu bilden, und nicht die anderen Zweige von Pflanze A besprühen, um eine Kontrollgruppe zu bilden.Machen Sie dasselbe für B und C.
Technische Replikate (Technische Replikate): Es handelt sich um ein wiederholtes Experiment, das darauf abzielt, Fehler zu vermeiden, die durch den Betrieb verursacht werden, bei dem es sich tatsächlich um ein doppeltes Loch im selben Material handelt.Sowohl Behandlungen als auch Kontrollen müssen über Replikationseinstellungen (mindestens drei) des Zielgens und des internen Referenzgens verfügen.
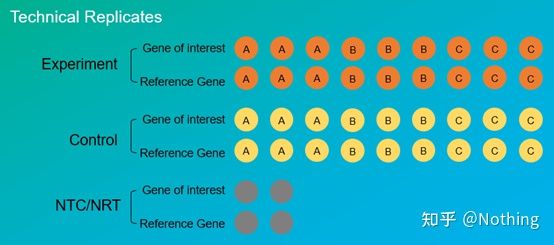
Technische Wiederholung
Nehmen wir als Beispiel noch einmal den mit Pestiziden behandelten Pfeffer.Für die Versuchsgruppe der Pflanze A haben wir drei PCR-Löcher von 1, 2 und 3 für ihr Zielgen bzw. ihr internes Referenzgen erstellt, um nach dem Nachweis den Durchschnitt zu ermitteln.Für die Kontrolle von Anlagen A werden Gruppen ebenfalls in gleicher Weise behandelt.Führen Sie die gleiche Behandlung auch für B- und C-Pflanzen durch.Das ist technische Wiederholung.
Das ist erwähnenswertWas in die Statistik einfließt, ist die biologische Wiederholung, und die technische Wiederholung dient dazu, zu testen, ob es zufällige Phänomene im experimentellen Prozess gibt, um die experimentellen Ergebnisse glaubwürdig zu machen, das heißt, um Fehler zu vermeiden, indem man ihren Durchschnitt bildet, wie wir oft sagen.
Negativkontrollen – NTC und NRT
NTC (No-Template Control), eine Kontrolle ohne Vorlage, wird verwendet, um zu überprüfen, ob das Versuchsmaterial kontaminiert ist.Im Allgemeinen wird Wasser als Vorlage verwendet.Kommt es zu einer fluoreszierenden Reaktion, ist dies ein Hinweis darauf, dass im Labor eine Nukleinsäurekontamination stattgefunden hat.
Diese Verschmutzungen entstehen durch: unreines Wasser, unqualifizierte Reagenzien, die endogene DNA enthalten, Primerverschmutzung, Verschmutzung von Laborgeräten, Aerosolverschmutzung usw., es müssen RNase-Scavenger und RNase-Inhibitoren verwendet werden.Aerosolverschmutzung ist am schwierigsten zu finden.Stellen Sie sich vor, dass Ihr Labor wie Smog ist und verschiedene Nukleinsäuren in der Luft schweben.

NRT (No-Reverse Transkriptase), die Kontrolle ohne reverse Transkription, ist die nicht revers transkribierte RNA als Negativkontrolle, also die Kontrolle der gDNA-Reste.
Bei der Genexpression wird die RNA-Menge durch den Nachweis der cDNA-Menge nach der reversen Transkription ermittelt.Wenn bei der RNA-Reinigung gDNA-Rückstände vorhanden sind, führt dies zu Fehlern in den experimentellen Ergebnissen, da es sich bei den tatsächlich erhaltenen Ergebnissen um gDNA und cDNA handelt.Auf Aggregatebene muss bei der RNA-Extraktion nicht nur cDNA, sondern auch gDNA vollständig entfernt werden.
MIQE (2) – Beispielinformationen
Die sogenannten Probeninformationen bedeuten, dass wir bei der Veröffentlichung eines Artikels über qPCR die Probeninformationen klar erläutern müssen, was ein unverzichtbarer Bestandteil des Artikels ist.Ebenso müssen wir bei der Verarbeitung von Proben auch unsere eigenen Abläufe regulieren, um die Gültigkeit der Proben sicherzustellen.

Die Beschreibung der Probe ist nur ein Ergebnis, und wir sollten den während des gesamten Experiments entnommenen Materialien mehr Aufmerksamkeit schenken.
Auswahl experimenteller Materialien
Blutproben – wählen Sie frisches Blut, nicht länger als 4 Stunden.Zellproben – Sammeln Sie frische Zellen in einer Phase kräftigen Wachstums.Tierisches Gewebe – Wählen Sie frisches, kräftig wachsendes Gewebe.Pflanzengewebe – Wählen Sie frisches, junges Gewebe.
Sie haben sicher bemerkt, dass in diesen wenigen Sätzen ein Schlüsselwort steckt: frisch.
Für die oben genannten Proben ist das Kit von Foregene das beste, kostengünstigste und stabilste Kit auf dem Markt, mit dem sich ihre DNA und RNA schnell und einfach extrahieren lassen.
Zell-Gesamt-RNA-Isolierungskit
Tier-Gesamt-RNA-Isolierungskit
Kit zur Isolierung pflanzlicher Gesamt-RNA
Plant Total RNA Isolation Kit Plus
Lagerung von Versuchsmaterialien
Generell raten wir davon ab, Proben aufzubewahren, sofern die Bedingungen dies zulassen.Es gibt jedoch viele Freunde, die nicht sofort nach der Probenahme Experimente durchführen können, und einige müssen sogar Tanks mit flüssigem Stickstoff zur Probenahme auf das Feld tragen.
Als fleißiger Freund kann ich nur sagen, dass Sie sich mit Reagenzien-Verbrauchsmaterialien nicht auskennen.Mittlerweile stellen viele Hersteller von Reagenzienverbrauchsmaterialien Reagenzien her, mit denen RNA-Proben bei Raumtemperatur gelagert werden können, und Sie können diese verwenden.Die herkömmliche Speichermethode ist die Lagerung von flüssigem Stickstoff, wobei ein kleiner Flüssigstickstofftank verwendet wird, der leicht zu transportieren ist.Nachdem Sie die Probe ins Labor zurückgebracht haben, lagern Sie sie in einem Kühlschrank bei -80 °C.

Für Experimente mit RNA muss das Sechs-Wörter-Prinzip befolgt werden:niedrige Temperatur, keine Enzyme,Undschnell .
Das Konzept der niedrigen Temperatur ist leicht zu verstehen;Ohne Enzyme ist RNase überall auf der Welt, in der wir leben, vorhanden (sonst wäre man durch HIV getötet worden). Daher ist es ein sehr wichtiges Konzept, wie man RNase bei Experimenten vermeidet.schnell,Es gibt kein Kung Fu auf der Welt, das nicht gebrochen werden kann, nur die Geschwindigkeit kann nicht gebrochen werden.
Daher gilt gewissermaßen: Je kürzer die Extraktionszeit, desto besser das Kit.Warum tutVorgeneDie Ausrüstung von s legt Wert auf Geschwindigkeit, weil sie sich damit gut auskennen.
PS: Manche Mädchen experimentieren sehr sorgfältig, aber nach mehreren Jahren Arbeit sind sie nicht so gut wie ein Slam Dunk.Sie empfinden Gott als ungerecht, beschweren sich über andere und streben nach Leben.Tatsächlich verstand sie es nicht.Er schützte die RNA nicht gut und der Slam-Dunk-Spieler war flink.Als er das Experiment durchführte, dachte er, dass er den Slam Dunk mit drei Malen, fünf Malen und zwei Divisionen beenden würde, aber er machte das Experiment gut.
Notiz: Langsamer, größere Wahrscheinlichkeit einer RNase-Invasion.Wie trainiere ich, schnell zu sein?Es gibt keine Möglichkeit, einfach mehr üben.
Für unterschiedliche Experimente und unterschiedliche Proben ist es dennoch erforderlich, mehr Literatur zu lesen und eine geeignete Methode für die Verarbeitung auszuwählen.Für den Prozess der Probenentnahme und -speicherung verlangt MIQE, dass dies klar in der Arbeit angegeben sein muss, damit die Gutachter die Zuverlässigkeit der Arbeit überprüfen können und es auch für die verblüfften Jugendlichen bequem ist, Ihr Experiment zu wiederholen.
Obwohl biologische Experimente schwierig sind, sind sie doch anspruchsvoll.Wenn Sie nicht aufpassen, können Sie die Welt umstürzen.Zum Beispiel SARS zu einer biochemischen Krise machen oder Hybridreis herstellen, um 1,3 Milliarden Menschen zu retten.Das Bild unten ist ein chemisches Experiment. Sie sollten verstehen, wie stolz Sie auf Ihre Forschung sind, wenn Sie nur sein schwanzartiges Aussehen betrachten.Vergiss es, schwärz ihn nicht.

MIQE (3) – Nukleinsäureextraktion.
Die Extraktion von Nukleinsäuren ist ein großes Ereignis, und alle molekularbiologischen Experimente beginnen mit der Extraktion von Nukleinsäuren.Lassen Sie uns zunächst den Inhalt von MIQE zur Nukleinsäureextraktion kopieren.
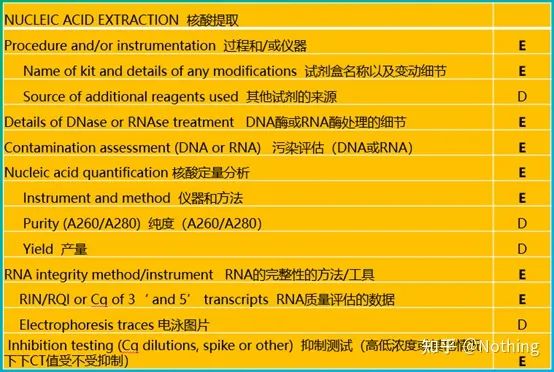
Wenn man diese Form betrachtet, kann man nicht an der Oberfläche bleiben.Die Form ist ein Dogma.Um ein Top-Student zu sein, muss man sich fragen, warum.Der wesentliche Inhalt dieser Tabelle ist: Verfolgendie Reinheit, Integrität, Konsistenz und Extraktionsmenge der RNA .
Der erste Teil desDer Prozess oder das Instrument ist der Schritt der Nukleinsäureextraktion.Wenn Sie zum Extrahieren einen automatischen Nukleinsäureextraktor verwenden (für Fortgeschrittene, kontaktieren Sie mich bitte zum Kauf), müssen Sie den Modellnamen des Instruments angeben.

Der Name des Kits und
Welches Kit für die Änderung verwendet wurde, welche speziellen Reagenzien hinzugefügt wurden oder welche speziellen Vorgänge durchgeführt wurden, sollte klar erklärt werden, damit andere Ihr Experiment problemlos wiederholen können.
Manche Leute fügen bei der Extraktion spezieller Proben einige spezielle Reagenzien hinzu, weil sie denken, dass dies ihre Geheimwaffe ist, und erzählen es anderen nicht.Während sie es geheim halten, verlieren sie gleichzeitig die Möglichkeit, Ihren Artikel zum Leuchten zu bringen.Seien Sie nicht schlau, Sie müssen ehrlicher sein als der alte Landsmann Zhang in der wissenschaftlichen Forschung. Wenn Sie schlau sein wollen, wird Sie der Artikel dumm machen.
Sie müssen sich die Produktnummer des Kits merkenwenn Sie das Kit bestellen und den Artikel schreiben.Auf dem Kit befinden sich im Allgemeinen zwei Nummern: Cat – Katalognummer (Produktnummer, Artikelnummer), Lot – Produktchargennummer (wird verwendet, um anzugeben, aus welcher Charge das Produkt stammt).
Darüber hinaus wird die CAS-Nummer häufig bei der Bestellung biochemischer Reagenzien verwendet, und ich werde sie gemeinsam bekannt machen.Die CAS-Nummer ist die Nummer, die die American Chemical Society jedem neuen chemischen Arzneimittel vergibt.Im Allgemeinen werden drei Zahlen durch einen Bindestrich verbunden.Rushuis CAS-Nummer: 7732-18-5.Chemikalien haben oft mehrere Aliase, aber die CAS-Nummer ist eindeutig.Wenn Sie ein Arzneimittel bestellen, können Sie zunächst dessen CAS-Nummer überprüfen.
Warum müssen wir diese Dinge in der näheren Umgebung klar beschreiben?Tatsächlich dient es auch dazu, die Qualität der RNA-Extraktion zu überprüfen.Durch den Einsatz von Instrumenten und Kits wird die RNA-Extraktion konsistenter.Der Extraktionsumfang gewöhnlicher Labore ist nicht groß und kann mit Kits erhalten werden.
Die Details der DNase- oder RNase-Behandlung
Das wichtige Problem der quantitativen Fluoreszenz-PCR besteht darin, eine DNA-Kontamination zu verhindern und bei einer Kontamination nicht zu experimentieren.Daher ist es unbedingt erforderlich, den Prozess anzugeben, mit dem Sie die DNA verarbeitet haben, um nachzuweisen, dass die DNA im experimentellen Prozess vollständig und vollständig entfernt wurde.dargestellt durch ein schematisches Diagramm.
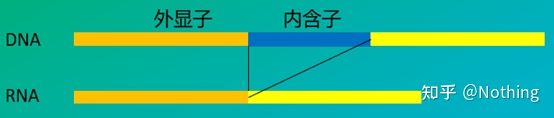
Schematische Darstellung von RNA und DNA
Im Allgemeinen besteht die Methode zur DNA-Entfernung darin, die RNA nach der Extraktion mit DNase zu behandeln.Allerdings handelt es sich hierbei um relativ alte Methoden.Kommerzielle RNA-Extraktionskits konnten DNA während des Extraktionsprozesses ohne Zugabe von DNase entfernen.Zum Beispiel eine Reihe von Bausätzen von Foregene.
Notiz: Das Entfernen von DNA während der RNA-Extraktion ist ein sehr gefährliches zweischneidiges Schwert, das die Operationszeit der RNA-Extraktion verlängert und das Risiko des RNA-Abbaus erhöht.Im Grunde handelt es sich um einen Kompromiss zwischen RNA-Ausbeute und Reinheit.
Darüber hinaus ist die Menge an DNase, die der Silica-basierten Adsorptionssäule zugesetzt wird, sehr gering und es muss hochwertige DNase verwendet werden, um den Effekt zu erzielen.Nicht optimierte DNase kann nicht schnell und vollständig verdaut werden.Dies ist ein Test des technischen Niveaus des Händlers.Natürlich gibt es noch seltsamere Händler, die damit prahlen, dass DNA ohne DNase entfernt werden kann.Man kann sagen, dass jeder, der damit prahlt, dass DNA ohne DNase vollständig entfernt werden kann, ein Hooligan ist.DNA ist eine relativ stabile doppelsträngige Struktur und kann nicht einfach durch Reden und Lachen ausgelöscht werden.
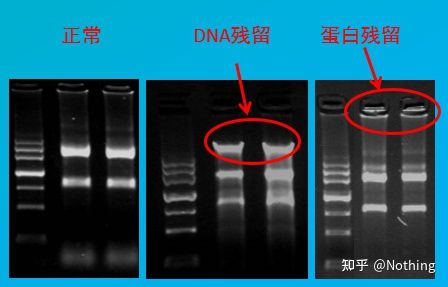
Kontaminationsbewertung
Bewertungsmethode: Elektrophorese-Detektion, 1 % Agarose, 6 V/cm, 15 Min., Beladung 1–3 µl
Quantitative Analyse von Nukleinsäuren
wird üblicherweise mit einem UV-Spektrophotometer gemessen.Lassen Sie mich zunächst die Bedeutung der drei Werte OD260, OD280 und OD230 bekannt machen.
·OD260nm: Dies ist die Absorptionswellenlänge des höchsten Absorptionspeaks der Nukleinsäure, und der beste gemessene Wert liegt zwischen 0,1 und 1,0.Wenn nicht, verdünnen oder konzentrieren Sie die Probe, um sie in den zulässigen Bereich zu bringen.
·OD280nm: Dies ist die Absorptionswellenlänge des höchsten Absorptionspeaks von Proteinen und phenolischen Substanzen.
·OD230nm: Dies ist die Absorptionswellenlänge des höchsten Absorptionspeaks von Kohlenhydraten.
Lassen Sie uns als Nächstes über die Rolle jedes einzelnen Indikators sprechen.Für A260 kann es zur Messung der Nukleinsäureausbeute verwendet werden.Wenn OD260=1, dsDNA=50μg/ml, ssDNA=37μg/ml, RNA=40μg/ml.
Für die Reinheit müssen wir uns die Verhältnisse ansehen, die wir üblicherweise sehen: OD260/280 und OD260/230.
·Reine DNA: OD260/280 entspricht ungefähr 1,8.Wenn er größer als 1,9 ist, deutet dies auf eine RNA-Verunreinigung hin, und wenn er kleiner als 1,6 ist, deutet dies auf eine Protein- und Phenolverunreinigung hin.
·Reine RNA: 1,7
·OD260/230: Ob DNA oder RNA, der Referenzwert beträgt 2,5.Wenn der Wert unter 2,0 liegt, bedeutet dies, dass Zucker, Salz und organische Stoffe verunreinigt sind.
RNA-Integrität
Es ist sehr wichtig, die Integrität der RNA zu messen.Im Allgemeinen ist es notwendig, ein RNA-Denaturierungsgelexperiment durchzuführen, um zu überprüfen, ob die Helligkeit zwischen 28S- und 18S-RNA eine zweifache Beziehung darstellt.Wenn die dritte Bande 5S erscheint, bedeutet dies, dass die RNA mit dem Abbau begonnen hat, außer bei Wirbellosen.

Daten zur RNA-Qualitätsbewertung: Zusätzlich zu den oben genannten Tests gibt es auch einige fortgeschrittenere Instrumententests im Hinblick auf die RNA-Integrität, wie beispielsweise den RQI-Integritätstest des automatischen Elektrophoresesystems Experion, der erkennen kann, ob RNA unsichtbar abgebaut wird.
In der wissenschaftlichen Forschung ist die quantitative Fluoreszenz-PCR ein Vergleich zwischen dem Zielgen und dem internen Referenzgen.Daher besteht das Hauptziel bei der Konservierung von RNA-Proben, der RNA-Extraktion usw. darin, die Integrität der RNA sicherzustellen.
Wie sich die Integrität der RNA auf das Gleichgewicht zwischen dem Zielgen und dem internen Referenzgen auswirkt, lässt sich anhand der folgenden Abbildung leicht verstehen.Der Abbau führt zu einer Unvollständigkeit des Gens. Unabhängig davon, ob es sich um die Unvollständigkeit des internen Referenzgens oder um die Unvollständigkeit des Zielgens handelt, hat dies große Auswirkungen auf die Daten.

Schematische Darstellung von Zielgen und Referenzgen, darf nicht wahr sein
Hemmungstest (ob der CT-Wert bei hoher oder niedriger Konzentration oder anderen Bedingungen unterdrückt wird)
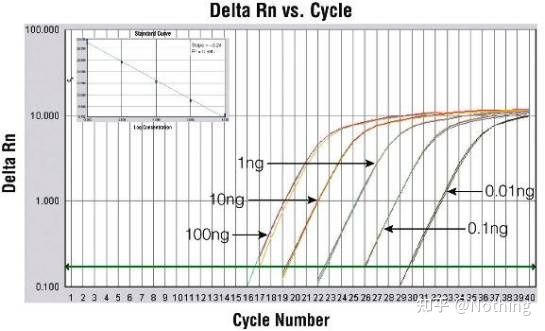
Am Beispiel dieser Abbildung lauten die Ct-Werte der fünf Kurven wie folgt.Die Verteilung der CT-Werte zwischen den Kurven ist ungleichmäßig und die Ct-Werte sind bei hohen und niedrigen Konzentrationen verzögert, was bei der PCR-Hemmung der Fall ist.
Kernpunkt: Im Prozess der RNA-Extraktion müssen wir Missverständnisse aufgeben und richtige festlegen.
Die falsche Vorstellung ist: Bei der RNA-Extraktion geht es nur um die Ausbeute und man denkt, je größer die gewonnene RNA-Menge, desto besser.Tatsächlich benötigen wir bei der Quantifizierung nicht viel RNA, wenn die Anzahl der Gene nicht sehr groß ist.Die Menge an RNA, die Sie extrahieren, ist mehr als ausreichend.
Das richtige Konzept ist:Bei der RNA-Extraktion sollte auf Reinheit, Integrität und Konsistenz geachtet werden.Durch Reinheit kann sichergestellt werden, dass die anschließende Reverse Transkription nicht gehemmt wird und die Daten nicht durch DNA beeinträchtigt werden.Integrität stellt das Gleichgewicht zwischen Zielsequenzen und internen Referenzen sicher.Konsistenz sorgt für eine stabile Probenladung.
MIQE (4) – Reverse Transkription
Missverständnis: das Streben nach einem höheren Probenvolumen.
Richtiges Konzept: Streben Sie nach Konsistenz (Stabilität). Unabhängig von der Menge der geladenen RNA bleibt die Effizienz der reversen Transkription konsistent und stellt sicher, dass Unterschiede in der cDNA tatsächlich Unterschiede in der mRNA widerspiegeln können.
Wir erklären diesen Vorgang anhand einer schematischen Darstellung:
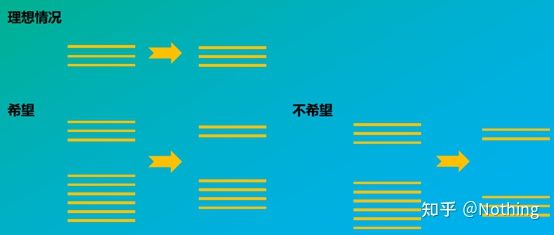
Das schematische Diagramm der Reverse-Transkriptions-Effizienz ist nicht wahr
Zunächst müssen wir den Unterschied zwischen dem Reverse-Transkriptionsprozess und dem PCR-Prozess verstehen.Die PCR durchläuft mehrere Erhitzungs- und Annealing-Prozesse und das Zielfragment wächst exponentiell;Während die umgekehrte Transkription diesen Prozess nicht hat, können wir uns vorstellen, dass die umgekehrte Transkription tatsächlich eins zu eins ist. Während des Replikationsprozesses werden so viele RNA-Stücke verarbeitet
Wie viele cDNA-Informationen erhalten werden können, sollte inzwischen verstanden werden, da große und kleine Fragmente revers transkribiert wurden und es unmöglich ist, sich auf ein Fragment zu konzentrieren.Und da die Menge an RNA relativ gering ist, ist auch die Menge an gewonnener cDNA relativ gering, im Gegensatz zur PCR, die einen Verstärkungseffekt hat, sodass ein Nachweis grundsätzlich unmöglich ist.
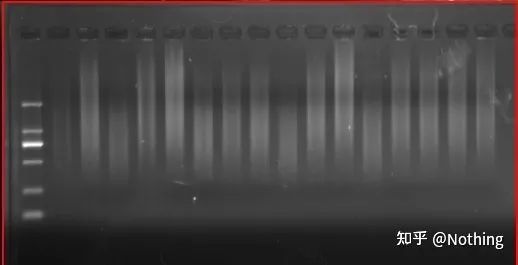
Ergebnisse der cDNA-Elektrophorese
Zweitens wird die Reverse Transkription im Idealfall eins zu eins durchgeführt, aber keine Reverse Transkriptase eines Unternehmens kann diesen Effekt erzielen.Grundsätzlich liegt die Effizienz der meisten Reverse Transkriptasen zwischen 30 und 50 %.Wenn dies der Fall ist, hätten wir lieber eine relativ stabile Reverse-Transkriptions-Effizienz, was wir in der Abbildung sehen wollen: 3 RNAs erhalten 2 cDNAs, 6 RNAs erhalten 4 cDNAs, also ist die Reverse-Transkriptions-Effizienz unabhängig von der geladenen Probe relativ stabil.Wir möchten keine Situation erleben, in der die Effizienz der reversen Transkription instabil ist und eine hohe Konzentration gehemmt wird.
Wie kann man also überprüfen, ob die Effizienz der reversen Transkription stabil ist?Die Methode ist sehr einfach, Sie müssen nur einen Vergleichstest durchführen: Einer besteht darin, nach der doppelten Verdünnung der RNA eine umgekehrte Transkription in cDNA durchzuführen, und die andere besteht darin, nach der umgekehrten Transkription in cDNA eine doppelte Verdünnung vorzunehmen und dann eine qPCR durchzuführen, um die erhaltene Steigung zu sehen. Ist sie konsistent?Als Top-Schüler sollten Sie es in Sekundenschnelle verstehen.Wie nachfolgend dargestellt:
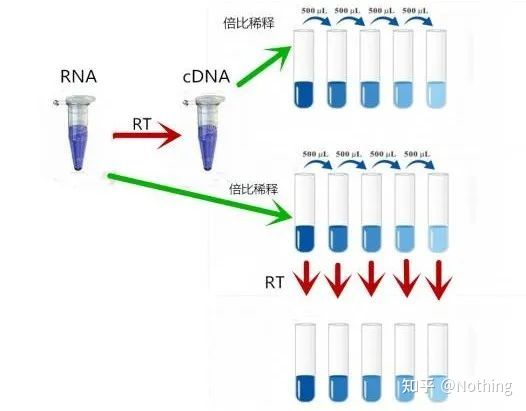
Verdünnung von RNA und cDNA, um zu testen, ob die Effizienz der reversen Transkription stabil ist
Reverse Transkriptase und Kit
Wie kann eine perfekte quantitative Fluoreszenz-PCR mit hervorragender Reverser Transkriptase und einem Kit ausgestattet werden?Reverse Transkriptase wird je nach Quelle grob in zwei Typen unterteilt: AMV oderM-MLV, und ihre Leistung ist die gleiche wie in der Tabelle.
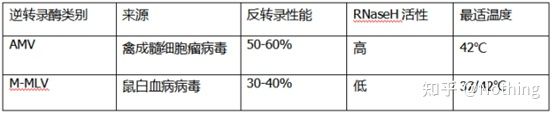
RNase H-Aktivität
RNase H ist Ribonuklease H, der chinesische Name ist Ribonuklease H, eine Endoribonuklease, die RNA in der DNA-RNA-Hybridkette spezifisch hydrolysieren kann.RNase H kann die Phosphodiesterbindungen in einzelsträngiger oder doppelsträngiger DNA oder RNA nicht hydrolysieren, d. h. sie kann einzelsträngige oder doppelsträngige DNA oder RNA nicht verdauen.Wird häufig bei der Synthese des zweiten cDNA-Strangs verwendet.
Es ist eine seltsame Sache.Wir sagen, dass Reverse Transkriptase RNase H-Aktivität aufweist, nicht dass Reverse Transkriptase RNase H enthält, und dass es möglicherweise nicht möglich ist, RNase H von Reverse Transkriptase zu trennen, möglicherweise aufgrund der Konformation bestimmter Gruppen in Reverse Transkriptase. Diese Aktivität wird durch die Reverse Transkriptase verursacht.
Daher verringert seine RNase H-Aktivität unabhängig von der höheren Effizienz der reversen Transkription von AMV die Ausbeute an cDNA.Natürlich optimieren Reagenzienhersteller ihre Produkte ständig, um die RNase H-Aktivität in der Reversen Transkriptase so weit wie möglich zu eliminieren und so die Ausbeute an cDNA zu erhöhen.
Glühtemperatur
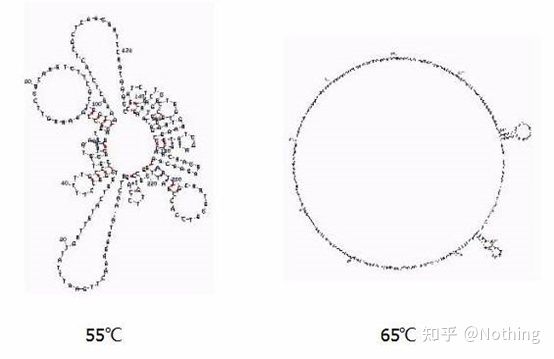
Sekundärstruktur der RNA bei verschiedenen Temperaturen
Sehen Sie sich die Abbildung oben für die Sekundärstruktur von RNA bei verschiedenen Temperaturen an und verwenden Sie das mFold-Online-Tool, um die Sekundärstruktur des Zielfragments unter bestimmten Temperatur- und Salzkonzentrationsbedingungen zu bestimmen.Bei 55 °C ist die Sekundärstruktur der RNA noch sehr komplex, die Reverse Transkriptase kann nicht funktionieren und die Sekundärstruktur kann erst bei 65 °C vollständig aufgelöst werden, während die optimale Temperatur von AMV und M-MLV weit unter dieser Temperatur liegt.
was zu tun ist?Die Sekundärstruktur ist die komplementäre Paarung der Matrize selbst, was zu einer starken Konkurrenz zwischen dem Primer und der Reverse Transkriptase und der Matrize führt, was zu einer Reihe von Problemen wie niedrigem E und schlechter Wiederholbarkeit führt.
was zu tun ist?Erhöhen Sie die Glühtemperatur nur so weit wie möglich.
Viele Reagenzienhersteller verbessern ihre Reverse Transkriptase durch Gentechnik.Einige erhöhen die Reaktionstemperatur, wie etwa Jifan und Aidelai, und andere entfernen die aktive Gruppe des RNase-H-Enzyms, um die Affinität zwischen dem Enzym und der RNA-Matrize zu verbessern.Eine hohe Affinität kann die Sekundärstruktur kompetitiv verdrängen und reibungslos durchlesen und außerdem die Effizienz der umgekehrten Transkription erheblich verbessern.
Kernpunkt: Reverse Transkription ist wichtiger, um die Konsistenz der Reverse-Transkriptions-Effizienz zu gewährleisten (Enzyme müssen nicht nur effizient, sondern auch stabil sein), als die Menge der geladenen Probe. Wenn es sich nicht um eine besonders groß angelegte fluoreszierende quantitative PCR handelt, ist sie überhaupt nicht möglich.Mehrere cDNAs.
Verschiedene Hersteller haben ebenfalls Anstrengungen unternommen, um die Konsistenz zu gewährleisten.Beispielsweise bieten die meisten Unternehmen die Reverse Transkription inzwischen als Standardpaket zum Verkauf an, was eine gute Wahl ist.
Zum Beispiel die RT Easy Series-Kits von Foregene:
RT Easy I (Master-Vormischung für Erststrang-cDNA-Synthese-Kit)
MIQE (5) – Zielgeninformationen

Die obige Abbildung erklärt
1. Ob dieses Gen für wiederholte Experimente wirksam ist, kann im Allgemeinen durch wiederholte Experimente überprüft werden.
2. Gen-ID, wissen Sie.
3. Genlänge, die Gesamtlänge des Zielgens ist definitiv kein Problem.Stellen Sie beim Entwerfen von Primern sicher, dass die Länge des Amplikons zwischen 80 und 200 bp liegt, um eine bessere Amplifikationseffizienz zu gewährleisten.
4. Informationen zum Sequenz-Blast-Vergleich: Das Zielgen muss in der Genbank verglichen werden, um eine unspezifische Amplifikation zu verhindern.
5. Vorhandensein von Pseudogenen.Ein Pseudogen ist eine DNA-Sequenz, die einem normalen Gen ähnelt, jedoch ihre normale Funktion verliert.Es kommt häufig in der Multigenfamilie der Eukaryoten vor.Es wird normalerweise durch ψ dargestellt.Es handelt sich um eine nicht funktionsfähige genomische DNA-Kopie im Genom, die der kodierenden Gensequenz sehr ähnlich ist.werden im Allgemeinen nicht transkribiert und haben keine klare physiologische Bedeutung.
6. Position der Primer relativ zu Exons und Introns.In den Anfangsjahren, als wir das Problem der DNA-Kontamination lösten, achteten wir häufig auf die Positionen von Primern, Exons und Introns und erwogen allgemein, Primer über Introns hinweg zu entwerfen, um eine DNA-Amplifikation zu vermeiden.Bitte sehen Sie sich die Abbildung unten an: Schwarz steht für Introns, verschiedene Blautöne stehen für Exons, Rosa steht für gemeinsame Primer und leuchtendes Rot steht für Intron-übergreifende Primer.
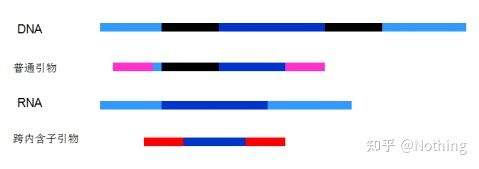
Schematisch, niemals wahr
Was für ein perfekter Plan das scheint, aber tatsächlich sind die Trans-Intron-Primer in den meisten Fällen nicht so magisch wie gedacht, und sie verursachen auch eine unspezifische Verstärkung.Der beste Weg, eine DNA-Kontamination zu verhindern, besteht also darin, die DNA vollständig zu entfernen.
7. Konformationsvorhersage.Verwenden Sie erneut dieses Beispiel und verwenden Sie das mFold-Online-Tool, um die Sekundärstruktur des Zielfragments bei einer bestimmten Temperatur und Salzkonzentration zu bestimmen.
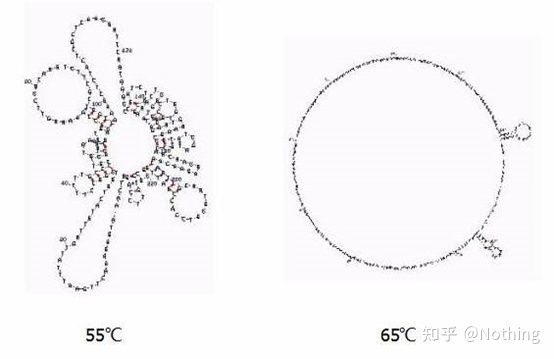
Sekundärstruktur der RNA bei verschiedenen Temperaturen
Die Sekundärstruktur ist die komplementäre Paarung der Matrize selbst, was zu einer starken Konkurrenz zwischen der Primer- und Matrizenpaarung führt und die Wahrscheinlichkeit einer Primerbindung geringer ist, was zu einer Reihe von Problemen wie niedrigem E und schlechter Wiederholbarkeit führt.Wenn es durch Softwarevorhersage kein Sekundärstrukturproblem gibt, wäre das großartig.Wenn dies der Fall ist, wird in unserem Folgeartikel speziell darauf eingegangen, wie dieses Problem gelöst werden kann.
MIQE (6) – qPCR-Oligonukleotide

Bei der quantitativen Fluoreszenz-PCR ist das erste, mit dem Sie jeden Tag zu kämpfen haben, die RNA-Extraktion, und das zweite, womit Sie möglicherweise mit dem Primer-Design zu kämpfen haben.
Zunächst prüfen wir noch die Regeln zur Grundierung anhand der MIQE-Checkliste.Es ist so einfach, dass die Drecksäcke lachen können, und wir können es in einem Satz beenden: Finden Sie die Sequenz und Position der Primersonde und die Modifikationsmethode heraus.Für die Primer-Reinigungsmethode ist die Primer-Synthese derzeit so günstig, dass qPCR einer PAGE und höheren Reinigungsmethoden würdig ist und die Informationen des Syntheseinstruments nicht wichtig sind.Viele Leute machen seit Jahrzehnten Grundierungen und wissen nicht, dass der Synthesizer ABI3900 ist.
Was die Prinzipien des Primer-Designs betrifft, müssen Sie sie nicht auswendig lernen, da die meisten Primer-Design-Software oder Online-Tools diese Probleme lösen können (empfohlenes Online-Tool primer3.ut.ee/) und 99,999 % des Primer-Designs nicht manuell durchgeführt werden. Schauen Sie, der Autor entwirft manchmal Hunderte von Primern pro Tag. Wenn Sie einen nach dem anderen lesen, wird er schielen.
Überprüfen Sie einfach die folgenden Punkte, nachdem die Primer entworfen wurden:
1. Entwerfen Sie Primer in der Nähe des 3′-Endes: Im Falle der Verwendung von Oligo-dT-Primern für die cDNA-Erststrangsynthese müssen die entworfenen Primer unter Berücksichtigung der Effizienz der reversen Transkription und der RNA-Integrität nahe am 3′-Ende entworfen werden, um die Amplifikationseffizienz zu verbessern.Erklären Sie es anhand eines Bildes wie folgt (das ist nicht verständlich):
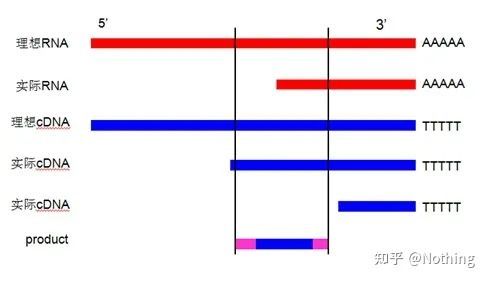
Warum sollten Primer nahe am 3′-Ende entworfen werden, das darf nicht wahr sein
2. TM-Wert: Der Tm-Wert liegt bei 55–65 °C (da die Exonukleaseaktivität bei 60 °C am höchsten ist), und der GC-Gehalt liegt bei 40 %–60 %.
3. BLAST: Um eine unspezifische Amplifikation des Genoms zu vermeiden, muss Blast zur ergänzenden Verifizierung eingesetzt werden.
MIQE(7) – qPCR-Prozess
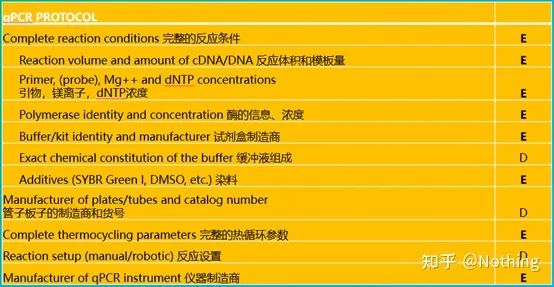
1. qPCR-Kit
Gemäß den Anforderungen von MIQE müssen wir im Artikel die vollständigen Reaktionsbedingungen klar beschreiben, einschließlich der Konfiguration des PCR-Reaktionssystems, des verwendeten Kits, des Herstellers, der Größe des Reaktionssystems, der Verwendung der Farbstoffmethode oder der Sondenmethode und der Einstellungen des PCR-Programms.Erfahrene Fahrer werden auf jeden Fall feststellen, dass bei der Auswahl des Kits die oben genannten Informationen im Wesentlichen festgelegt sind.
Derzeit ist die Herstellung und Produktion von fluoreszierenden quantitativen PCR-Kits eine sehr ausgereifte Technologie.Solange Sie sich nicht für extrem schlechte Hersteller entscheiden, ist die Wahrscheinlichkeit von Problemen nicht hoch, dennoch möchten wir Ihnen ein paar Punkte mitteilen:
Hot-Start-Taq-Enzym:Der wichtigste Teil der PCR ist das Hot-Start-Taq-Enzym.Die auf dem Markt erhältlichen Hot-Start-Enzyme werden im Allgemeinen in zwei Typen unterteilt: Der eine ist ein chemisch modifiziertes Hot-Start-Enzym (man kann es sich als Paraffin-Einbettung vorstellen) und der andere ist ein Hot-Start-Enzym zur Antikörpermodifikation (Antigen-Antikörper-Bindung).Chemische Modifikation ist eine frühe Möglichkeit, Enzyme heiß zu starten.Bei Erreichen einer bestimmten Temperatur setzt das Enzym seine Aktivität frei.Das antikörpermodifizierte Hot-Start-Enzym nutzt biologische Methoden, um die Aktivität des Enzyms zu blockieren.Bei Erreichen einer bestimmten Temperatur wird der Antikörper denaturiert und als Protein inaktiviert und die Enzymaktivität wird aktiviert.
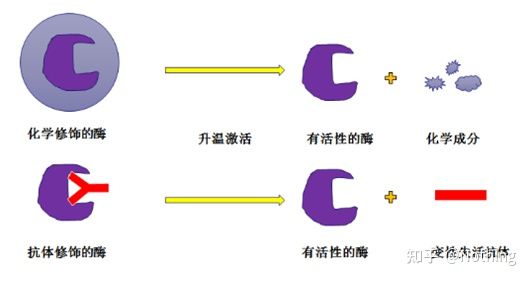
Doch welchen Nutzen hat das?Dies ist der Fall, die Freisetzungsaktivität von Antikörper-modifizierten Enzymen ist schneller als die von chemisch modifizierten Enzymen, sodass Antikörper-modifizierte Enzyme hinsichtlich der Empfindlichkeit einen leichten Vorteil haben, sodass in den auf dem Markt befindlichen Kits grundsätzlich keine chemisch modifizierten Enzyme enthalten sind.Wenn ja, dann steckt die Technologie dieses Herstellers immer noch im Zeitalter des Jahrtausends fest.
Magnesiumionenkonzentration:Die Magnesiumionenkonzentration ist bei der PCR-Reaktion sehr wichtig.Eine angemessene Magnesiumionenkonzentration kann die Freisetzung der Taq-Enzymaktivität fördern.Bei zu geringer Konzentration wird die Enzymaktivität deutlich reduziert;ist die Konzentration zu hoch, wird die enzymkatalysierte unspezifische Amplifikation verstärkt.Die Konzentration der Magnesiumionen beeinflusst auch das Annealing von Primern, die Schmelztemperatur der Matrize und der PCR-Produkte und beeinflusst dadurch die Ausbeute an amplifizierten Fragmenten.Die Konzentration der Magnesiumionen wird im Allgemeinen auf 25 mM kontrolliert.Für ein gutes Kit muss natürlich die Konzentration der Magnesiumionen gut kontrolliert werden.Einige Händler fügen dem Reagenz einen Magnesiumionen-Chelatbildner hinzu, der den Effekt einer automatischen Anpassung der Magnesiumionenkonzentration erzielen kann.
Konzentration des Fluoreszenzfarbstoffs:Der fluoreszierende Farbstoff, bei dem es sich um das von uns üblicherweise verwendete SYBR-Grün handelt, erzeugt Fluoreszenz hauptsächlich durch Bindung an die kleine Furche doppelsträngiger DNA, da die Bindung des Farbstoffs an doppelsträngige DNA unspezifisch ist, d.
PS: Aufgrund seiner lichtempfindlichen Eigenschaften werden die auf dem Markt erhältlichen Produkte in der Regel in braunen, undurchsichtigen Zentrifugenröhrchen verpackt (wie im Bild unten gezeigt).Dies wird jedoch auf ein Problem stoßen.Bei der Probenahme ist schwer zu erkennen, ob die Flüssigkeit angesaugt wird.In dieser Hinsicht ist Qingke tatsächlich am benutzerfreundlichsten (siehe Abbildung unten), und die transparente Tube ist in einem undurchsichtigen Blechbeutel verpackt.Legen Sie es dann in einen Blechbeutel und achten Sie darauf, Licht und Probenahme zu vermeiden.Sie müssen die richtige Produktnummer auswählen.TSE204 ist eine super kostengünstige Existenz, die mich dazu bringt, Gras zu pflanzen.

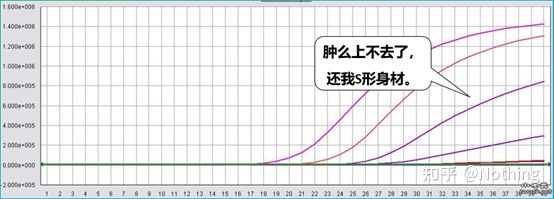
Auch die Konzentration des Fluoreszenzfarbstoffs ist sehr wichtig.Wenn die Konzentration zu niedrig ist, wird die Amplifikationskurve im späteren Stadium nicht ansteigen und ist nicht perfekt;Ist die Konzentration zu hoch, kommt es zu Störgeräuschen.Da die quantitative Fluoreszenz-PCR hauptsächlich vom CT-Wert abhängt, ist der Tiefpunkt besser als der Höchstpunkt, wenn die Konzentration des Fluoreszenzfarbstoffs nicht richtig eingestellt wird.Am besten ist natürlich die entsprechende Farbstoffkonzentration.
ROX: ROX-Farbstoffe werden zur Korrektur von Well-to-Well-Fluoreszenzsignalfehlern verwendet.Einige Gerätehersteller verlangen eine Kalibrierung, andere nicht.Beispielsweise erfordert die Verwendung des Echtzeit-PCR-Amplifikationsgeräts von Thermo Fisher Scientific in der Regel eine Kalibrierung, einschließlich 7300, 7500, 7500Fast, StepOnePlus usw. Die allgemeine Kit-Anleitung wird dies beschreiben.
Der qPCR-Mix von Foregene enthält außerdem ROX-Farbstoff, der sich praktisch für die Verwendung in verschiedenen Modellen eignet.
Behandlung schwacher Wasserstoffbrückenbindungen: Die Behandlung schwacher Wasserstoffbrückenbindungen ist eine relativ technische Angelegenheit.Ich habe die Handbücher vieler Kits nicht gelesen, aber keiner von ihnen erwähnte dieses Thema.Tatsächlich ist es so wichtig.Die Kombination der Basen hängt hauptsächlich von der Stärke der Wasserstoffbrückenbindungen ab.Starke Wasserstoffbrückenbindungen sind eine normale Verstärkung, und schwache Wasserstoffbrückenbindungen führen zu einer unspezifischen Verstärkung.Wenn schwache Wasserstoffbrückenbindungen nicht gut eliminiert werden können, ist eine unspezifische Verstärkung nicht zu vermeiden.Im Sinne des Autors ist dieses Problem nur wenigen Unternehmen aufgefallen.Beim Kauf des Bausatzes können Sie sich darüber informieren, ob Sie diesbezüglich eine Lösung für den Bausatz, für den Sie sich entscheiden möchten, in Betracht gezogen haben.
Reaktionsvolumen: Das 20-50ul-System wird häufiger verwendet und kleinere Volumina können wahrscheinlich Fehler verursachen.Im Allgemeinen wird in den Kit-Anweisungen die Verwendung von PCR-Reaktionsvolumina empfohlen.Seien Sie nicht schlau und verwenden Sie kleinere Mengen, um Kosten zu sparen.das Ziel von.Das von den Händlern empfohlene Volumen wurde tatsächlich getestet und es kann sein, dass sie das Problem der durch kleine Mengen verursachten Fehler nicht lösen können.
2. Der Hersteller und die Artikelnummer der Rohrplatte
Jeder kennt das Prinzip der fluoreszierenden quantitativen PCR.Die Fluoreszenzsammlung erfolgt hauptsächlich durch PCR-Röhrchendeckel.Achten Sie bei der Auswahl von PCR-Verbrauchsmaterialien auf zwei Punkte: gute Lichtdurchlässigkeit und Eignung für das Gerät.Im Allgemeinen sind die Boards und Röhren der Mainstream-Marken in Ordnung, aber Sie müssen hinsichtlich der Anpassung sorgfältig auswählen, sonst können Sie das Instrument nicht verwenden.

4. Wissen auf höchstem Niveau
MIQE (8) – qPCR-Validierung
Das ist die oberste Priorität von qPCR!So viele Helden sind hier in den Sand gefallen.Natürlich ist es auch möglich, dass Sie Glück haben und die Gene, die Sie untersucht haben, einfach sind, sodass Sie im Wind durch die Eishöhle schwebten.Die Verifizierungsinformationen von qPCR sollen die Zuverlässigkeit der Daten testen.Wir listen die erforderlichen Verifizierungsinformationen wie folgt auf:
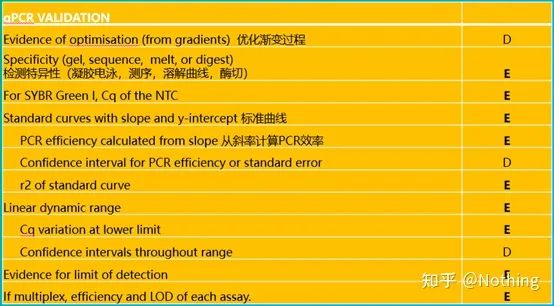
1. Spezifitätstest
Die Spezifität der Zielgenamplifikation wird getestet, indem überprüft wird, ob das Elektrophoresebild eine einzelne Bande darstellt;Sequenzverifizierung;Schmelzkurve, um zu sehen, ob die Peakkarte einzeln ist;Überprüfung der Enzymverdauung und andere Methoden.
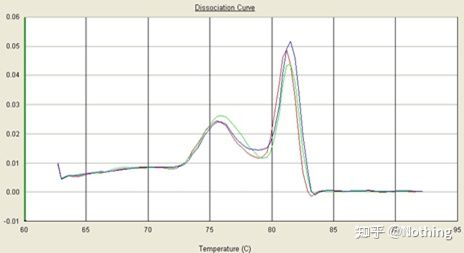
Hier konzentrieren wir uns auf tDie Analyse der unspezifischen Amplifikation mit der Methode der Schmelzkurven.Wenn wir Primer entwerfen, muss die Größe des Produktfragments im Allgemeinen im Bereich von 80–200 bp liegen, wodurch die Schmelztemperatur des PCR-Produkts im Bereich von 80–85 °C liegt.Wenn daher verschiedene Peaks vorhanden sind, müssen andere unspezifische Amplifikationsprodukte vorhanden sein.wenn der Peak unter 80 °C erscheint, wird er im Allgemeinen als Primer-Dimer betrachtet;Wenn der Peak über 85 °C erscheint, wird allgemein davon ausgegangen, dass es sich um eine DNA-Kontamination oder eine unspezifische Amplifikation großer Fragmente handelt.
Hinweis: Manchmal gibt es nur einen einzigen Peak bei 80 °C.Zum jetzigen Zeitpunkt muss an diesem Konzept festgehalten werden.Es ist wahrscheinlich, dass es sich bei den Amplifikationsergebnissen ausschließlich um Primer-Dimere handelt.
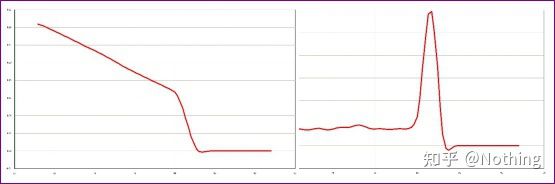
Normale Schmelzkurve (einzelner Peak ohne unspezifische Verstärkung)
Problematische Schmelzkurve (unspezifische Verstärkung von Störpeaks)
【Fall Analyse】
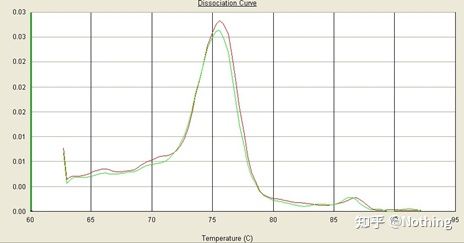
Es gibt einen Hauptpeak, aber das Primer-Dimer ist schwerwiegend
Die Single-Peak-Schmelzkurve in der Abbildung unten kann Ihre Augen leicht täuschen und denken, dass es sich um ein perfektes Experiment handelt, aber das Ergebnis ist völlig falsch.Zu diesem Zeitpunkt müssen wir uns die Schmelztemperatur ansehen.Die Spitzentemperatur liegt unter 80 °C, was vollständig Primer-Dimer ist.
Kein Zielfragment, alle Primer-Dimere
Hier kann mein Bruder nicht aufhören.Das Bild unten ist ein Foto, das mit einem Mobiltelefon aufgenommen wurde, das mir ein Drecksack geschickt hat.Die von ihm verwendeten Reagenzien sind allesamt gängige Marken in der Branche.Er wechselte von einer T-Präfix-Marke zu einer anderen T-Präfix-Marke.Ich denke, Sie haben es bereits erraten.Der Drecksack rief mir zu: „Das im ersten Bild verwendete Reagenz ist zu gut und der Peak ist einfach.“Später, nachdem Sie das von Ihnen empfohlene Reagenz verwendet haben, ähnelt es dem zweiten Bild mit gemischten Peaks.Du hast mich unglücklich gemacht.„
Trennen Sie die beiden Diagramme.Auf den ersten Blick hat einer einen einzelnen Peak und der andere einen doppelten Peak.Unsinn, ein einzelner Peak ist natürlich in Ordnung.Ist das wahr?
Schlimmer als Dou E. Wenn ich die beiden Bilder in das Bild unten füge, werden Sie es sofort verstehen.Tatsächlich lassen wir uns von solchen Bildern leicht lähmen.Nach sorgfältiger Analyse haben wir Folgendes herausgefunden: Der Peak der ersten Zahl liegt bei 75 °C, was vollständig Primer-Dimer ist;Der Peak der zweiten Zahl erscheint bei 75°C und 82°C, zumindest dort. Das Produkt erscheint.
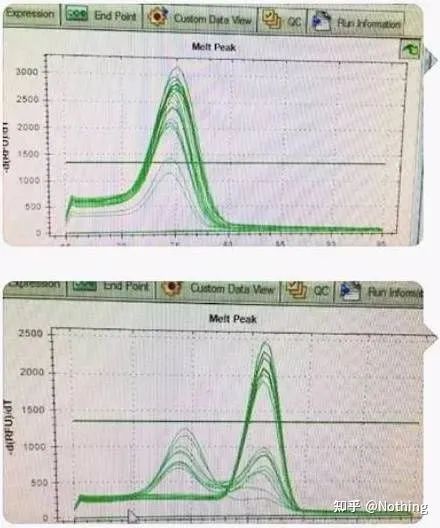
Bilder von Rückmeldungen von Studierenden
Das grundlegende Problem ist also nicht das Problem der Reagenzien, sondern das Problem des Primerdesigns.Gleichzeitig beweist es auch, dass einige große Marken nicht von Eisenqualität sind, und es beweist auch, was mein Bruder zuvor gesagt hat: Es ist nicht die Reagenzmarke, die Ihren Artikel unterstützt.Es ist Ihr Artikel, der die Marke der Reagenzien gestützt hat.Stellen Sie sich vor, wenn der Dreckskerl die Reagenzien nicht wechseln würde, würden die falschen Daten an das Journal gesendet, und was passieren würde, wäre eine Tragödie.
2. Ct-Wert der Blindkontrolle
Erklären Sie nicht, wenn die Blindkontrolle einen Ct-Wert hat, handelt es sich dann nicht um eine Verschmutzung?Sie müssen jedoch noch verstehen, welche Blindkontrolle einen Ct-Wert hat.Wenn es sich um NTC handelt, bedeutet dies, dass Fremd-DNA vorhanden ist, z. B. eine Reagenzverunreinigung.Wenn es sich um NRT handelt, bedeutet dies, dass die extrahierte RNA eine DNA-Kontamination aufweist.
3. Standardkurve
Unter Einbeziehung der Steigung und der Berechnungsformel kann die PCR-Effizienz anhand der Formel berechnet werden.Für ein perfektes Experiment muss sich die Steigung der Standardkurve 3,32 nähern und R² sich 0,9999 nähern.
4. Linearer Dynamikbereich
Der dynamische Bereich der Reaktion ist linear.Gemäß der zur Erstellung der Standardkurve verwendeten Vorlage sollte der dynamische Bereich mindestens 5 Konzentrationsgradienten umfassen und auf die Änderung der Ct-Werte bei hohen und niedrigen Konzentrationsgradienten achten.
5. Erkennungsgenauigkeit
Änderungen der qPCR-Ergebnisse, d. h. schlechte Wiederholbarkeit, d. h. schlechte Präzision, werden durch viele Faktoren verursacht, darunter Temperatur, Konzentration und Betrieb.Die qPCR-Präzision ist im Allgemeinen mit abnehmender Kopienzahl weniger kontrollierbar.Idealerweise handelt es sich um eine Variation innerhalb des Experiments. Diese technische Variation sollte sich von der biologischen Variation unterscheiden, und biologische Replikate können statistische Unterschiede in den qPCR-Ergebnissen zwischen Gruppen oder Behandlungen direkt berücksichtigen.Insbesondere für diagnostische Tests muss die beste Inter-Assay-Präzision (Wiederholbarkeit) über Standorte und Betreiber hinweg angegeben werden.
6. Nachweiseffizienz und LOD (in Multiplex-qPCR)
LOD ist die niedrigste Konzentration von 95 % der nachgewiesenen positiven Proben.Mit anderen Worten: Die in einem Satz von Zielgenreplikaten enthaltene LOD-Konzentration sollte 5 % der fehlgeschlagenen Reaktionen nicht überschreiten.Bei der Durchführung einer Multiplex-qPCR-Analyse, insbesondere für den gleichzeitigen Nachweis von Punktmutationen oder Polymorphismen, muss die Multiplex-qPCR den Nachweis erbringen, dass die Genauigkeit mehrerer Zielfragmente im selben Röhrchen nicht beeinträchtigt wird; Mehrfacherkennung und Einzelröhrchen-Detektion. Effizienz und LOD sollten gleich sein.Insbesondere wenn hochkonzentrierte Zielgene und niedrigkonzentrierte Zielgene gleichzeitig amplifiziert werden, muss dieses Problem beachtet werden.
Probleme und LösungenIm Allgemeinen konzentrieren sich die beim qPCR-Debugging häufig auftretenden Probleme auf die folgenden Aspekte:
·unspezifische Verstärkung
·Schwierige Wahl der Primerkonzentration und Probleme mit Primer-Dimeren
·Die Glühtemperatur ist ungenau
·Sekundärstruktur beeinflusst die Verstärkungseffizienz
unspezifische Verstärkung
unspezifische VerstärkungTritt auf, wird im Allgemeinen darüber nachgedacht, ob das Primer-Design nicht geeignet ist. Wenn Sie es jedoch nicht eilig haben, die Primer zu wechseln, können Sie zunächst die folgenden Methoden ausprobieren (das Prinzip ist ebenfalls beigefügt):
·Erhöhen Sie die Glühtemperatur – versuchen Sie, schwache Wasserstoffbrückenbindungen nicht mehr aufrechtzuerhalten;
·Verkürzen Sie die Glüh- und Verlängerungszeit – verringern Sie die Wahrscheinlichkeit schwacher Wasserstoffbrückenbindungen;
·Reduzieren Sie die Primerkonzentration – verringern Sie die Wahrscheinlichkeit der Bindung überflüssiger Primer und Nicht-Zielregionen;
Geringe Verstärkungseffizienz
Das Gegenteil zur unspezifischen Amplifikation – niedrige Amplifikationseffizienz – und die Maßnahmen zum Umgang mit niedriger Amplifikationseffizienz sind genau das Gegenteil:
·Verlängern Sie die Glüh- und Verlängerungszeit;
· Wechseln Sie zur dreistufigen PCR und reduzieren Sie die Annealing-Temperatur.
·Erhöhen Sie die Primerkonzentration;
Ps: Viele in den 90er Jahren geborene Doktoranden sind nicht bereit zu lernen, wie man Experimente debuggt, und hoffen, dass das Kit das Problem vollständig lösen kann (wenn Sie nach dem Abschluss zu einem Reagenzienunternehmen gehen möchten, um Forschung und Entwicklung zu betreiben).Um das Problem einfach zu lösen, müssen Dummköpfe immer noch die Einführung des Reagenzienherstellers lesen, um zu sehen, ob es einen Faktor gibt, der schwache Wasserstoffbrückenbindungen absorbiert.
Schwierige Wahl der Primerkonzentration und Probleme mit Primer-Dimeren
Methode 1: Im Allgemeinen enthalten die Kit-Anweisungen für qPCR empfohlene Systeme und empfohlene Primerkonzentrationen.
Methode 2: Debugging durch Einstellen des Primerkonzentrationsgradienten.Das Bild unten wurde zur Veranschaulichung von einem Unternehmen gestohlen.Die folgende Abbildung zeigt die quantitativen Fluoreszenzergebnisse, die mit drei Primerkonzentrationsgradienten (100 nM, 250 nM, 500 nM) und vier Templatkonzentrationsgradienten (0,1 ng, 1 ng, 10 ng, 100 ng) erzielt wurden.Der Ct-Wert der Versuchsergebnisse wird wie folgt aufgetragen:
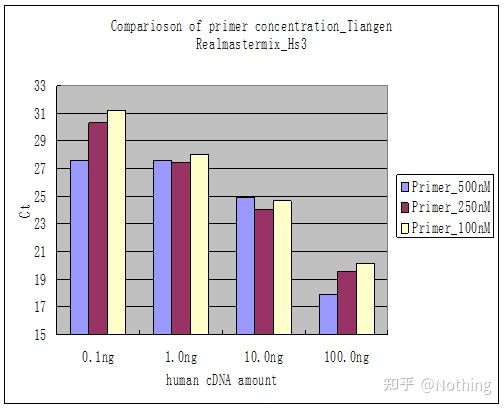
Auswahl der Primerkonzentration Verketten Sie jede Primerkonzentration wie folgt in einer Zeile:
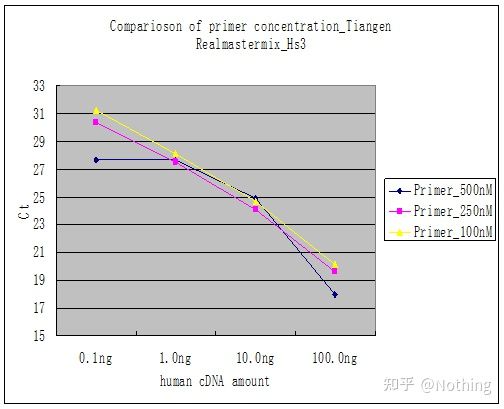
Die Wahl der Primerkonzentration ist offensichtlich, die lineare Beziehung der Primerkonzentration von 100 nM und 250 nM ist besser und die lineare Beziehung der Primerkonzentration von 500 nM ist relativ schlecht.Bei 100 nM und 250 nM ist der Ct-Wert von 250 nM relativ gering, sodass die optimale Primerkonzentration 250 nM beträgt.Generell sind in der Schmelzkurve starke Primer-Dimere zu erkennen.Was ist, wenn die entworfenen Primer Primer-Dimere nicht vermeiden können?
Methode 3: Reduzieren Sie die Menge an Primern und erhöhen Sie die Glühtemperatur (keine Erklärung erforderlich).
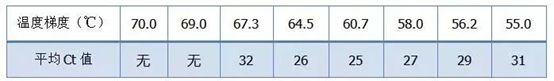
Der Erfahrungswert der Glühtemperatur beträgt 60°C.Wenn Sie sich nicht sicher sind, wie wählen Sie eine geeignetere Glühtemperatur aus?Die Antwort ist dieselbe wie bei der Wahl der Primerkonzentration –Gradiententest.Machen Sie ein Foto der Firma Bio-rad, um das Problem zu veranschaulichen.Stellen Sie für die Amplifikation eines bestimmten Zielfragments acht Temperaturgradienten mit jeweils drei Wiederholungen ein, und die erhaltene Amplifikationskurve sieht wie folgt aus:
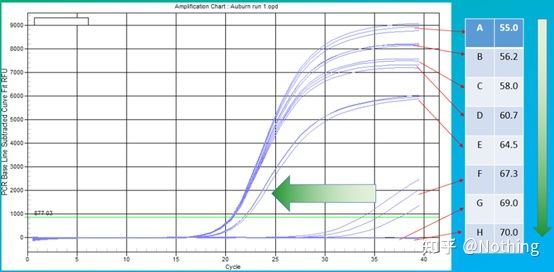
Auswahl der Glühtemperatur:
·70 °C, 69 °C – Grundsätzlich können die Primer nicht kombiniert werden, es findet also keine Amplifikation statt.
·67,3°C – Zu Beginn gibt es eine kleine Verstärkung und der Ct-Wert ist relativ groß.
·64,5°C – Der Ct-Wert nimmt ab.
·Bei 60,7 °C, 58,0 °C, 56,2 °C und 55,0 °C waren die Ct-Werte grundsätzlich stabil, die endgültigen Fluoreszenzwerte waren jedoch unterschiedlich.
Wie man wählt?Prinzip: Das erste Prinzip ist der höhere Ct-Wert.Wählen Sie für den gleichen Ct-Wert eine höhere Annealing-Temperatur, um Dimerisierung und unspezifische Amplifikation zu vermeiden.Obwohl es bei 55 °C einen höheren Fluoreszenzwert gibt, können Dimere oder unspezifische Verstärkung darin enthalten sein.
Aber wenn Sie so schlau sind wie Sie, werden Sie auf jeden Fall denken: Wenn die PCR-Reaktion sehr spezifisch ist, sollten die hohen und niedrigen Punkte logischerweise keine Wirkung haben, solange die Primerkonzentration die Mindestanforderung übersteigt, genau wie Fluoreszenzfarbstoffe und dNTPs.Solange die Glühtemperatur richtig optimiert wird, wird der Einfluss der Primerkonzentration auf den Ct-Wert natürlich minimiert.
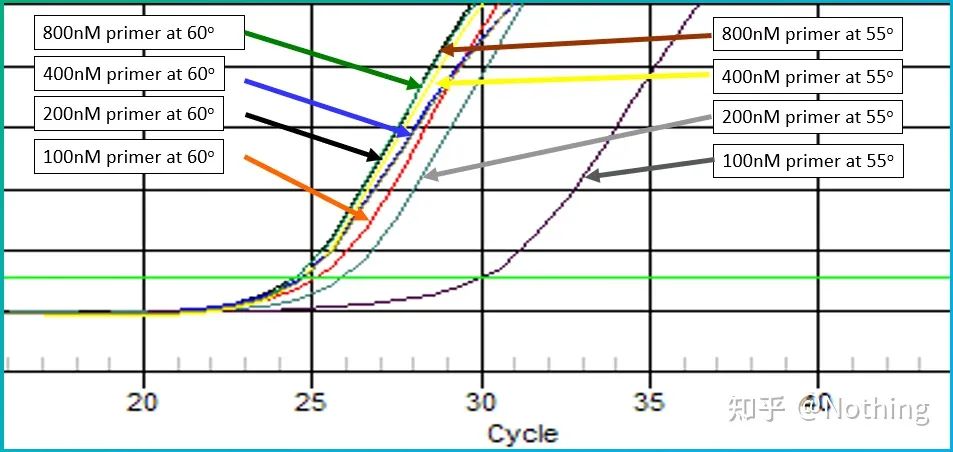
Die Glühtemperatur wird ordnungsgemäß optimiert und die Auswirkung der Primerkonzentration auf die CT wird minimiert
Die Sekundärstruktur beeinflusst die Verstärkungseffizienz
Nehmen wir das Bild von Bio-rad, um das Problem zu veranschaulichen.Außerdem wird ein Temperaturgradient entworfen, um ein Gen mit einer Sekundärstruktur zu verstärken.
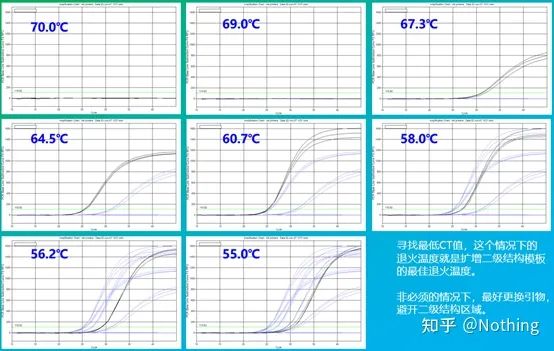
Es entsteht eine Sekundärstruktur
Es ist ersichtlich, dass mit abnehmendem Temperaturgradienten Produkte aufzutreten beginnen und der Ct-Wert nach vorne wandert und den Minimalwert bei 60,7 °C erreicht. Mit abnehmendem Temperaturgradienten wird der Ct-Wert dann größer.Umgekehrt öffnet sich mit steigender Temperatur die Sekundärstruktur und die Verstärkungseffizienz steigt.Nach Erreichen einer bestimmten Temperatur kann eine Erhöhung der Temperatur die Verstärkungseffizienz nicht mehr verbessern.Weil die Primer zu diesem Zeitpunkt nicht stabil kombiniert werden können.Deshalb,Suchen Sie nach der Temperatur mit dem niedrigsten Ct-Wert, was die beste Temperatur zur Verstärkung der Sekundärstrukturvorlage ist!Kluge Narren müssen natürlich wissen, dass es am besten ist, die Primer zu wechseln und den Sekundärstrukturbereich zu meiden, wenn dies nicht notwendig ist.
5. Anwendungsebene
MIQE – Datenanalyse

Die Datenanalyse erfolgt hauptsächlich mit dem fluoreszierenden quantitativen PCR-Gerät.Im vorherigen Artikel wurden viele Datenanalysearbeiten durchgeführt, beispielsweise die Blindkontrolle, die im Versuchsdesign erläutert wurde.Die internen Referenzgene, Wiederholungszahlen usw. wurden geklärt.Hier erklären wir hauptsächlich die Anwendung von qPCR.
qPCR ist weit verbreitet und die experimentelle Verifizierung und Nukleinsäurediagnose sind die am häufigsten verwendeten Szenarien.
absolute Quantifizierung
Log (Anfangskonzentration) hat eine lineare Beziehung zur Anzahl der Zyklen.Eine Standardkurve kann aus einem Standard mit bekannter anfänglicher Kopienzahl erstellt werden, d. h. es kann die lineare Beziehung der Amplifikationsreaktion ermittelt werden.Anhand des Ct-Wertes der Probe kann die Konzentration in der Probe berechnet werden.Die Anzahl der einzuschließenden Vorlagen.
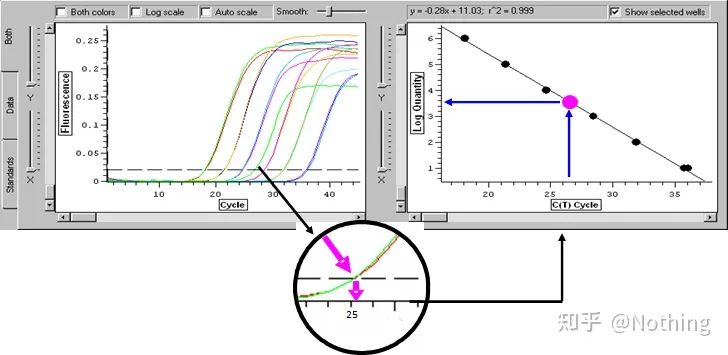
Absolute quantitative Berechnungsmethode
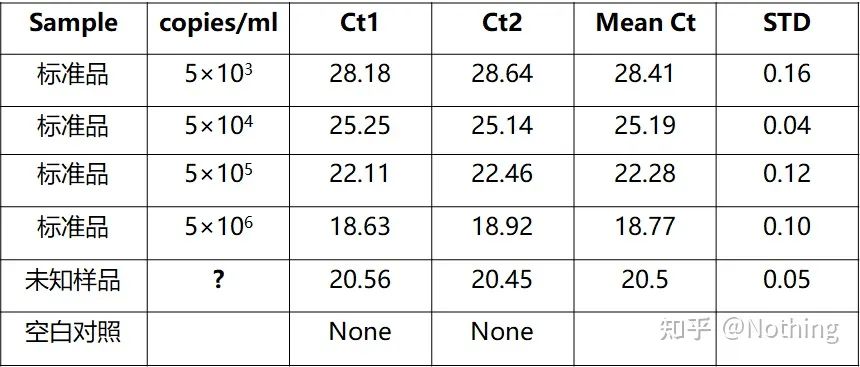
Die absolute Quantifizierung muss auf der Standardkurve basieren.Um eine Standardkurve zu erstellen, ist ein Standard erforderlich.Normalerweise ist der Standard ein Plasmid, das durch Klonierung des Zielgens gewonnen wird.Warum ist es ein Plasmid?Weil zirkuläre Plasmid-DNA am stabilsten ist.Verdünnen Sie das Standardprodukt in 5 bis 6 Steigungen entsprechend dem Verdopplungsverhältnis (10-fache Verdünnung) und achten Sie beim Verdünnen auf Gleichmäßigkeit.Lassen Sie den Ct-Wert zwischen 15 und 30 liegen.
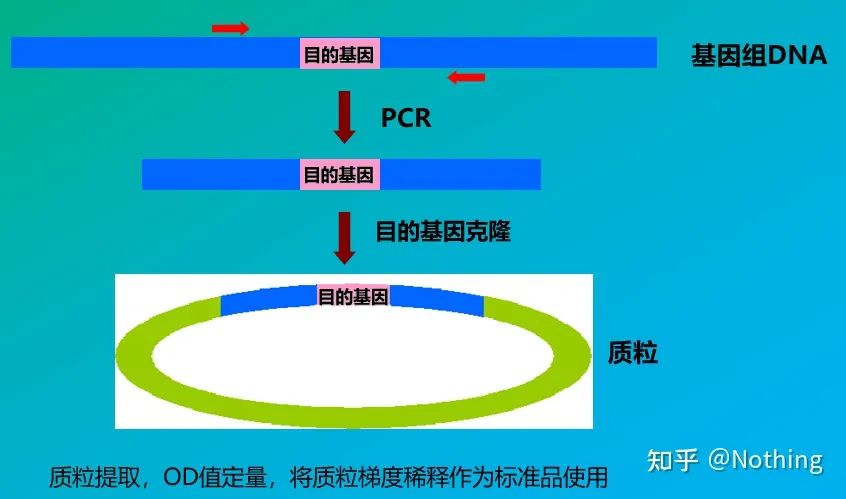
Standardvorbereitung
Gleichzeitig sollte auch die zu untersuchende Probe entsprechend verdünnt werden (Verdünnungsfaktor beachten) und der Ct-Wert sollte ebenfalls zwischen 15 und 30 liegen.Das Standardprodukt und die zu prüfende Probe werden zusammen auf die Maschine gegeben.Nach dem Lauf wurde eine Standardkurve mit der Standardsubstanz erstellt und die zu untersuchenden Proben zur Berechnung der Konzentration in die Standardkurve gebracht.
Die Hepatitis-B-Virus-HBV-Quantifizierung ist eine typische absolute Quantifizierung, mit der die Viruskopienzahl in 1 ml Blut berechnet werden kann.
Berechnung der Exemplarzahl
Zu testende Probenkonzentration (ng/ul) = OD260 × 50ug/ml × Verdünnungsfaktor
Molekulargewicht der Probe = Anzahl der Basen × 324
Die Kopienzahl der zu testenden Probe (Kopien/ul) = die Konzentration der zu testenden Probe / das Molekulargewicht der Probe × 6 × 1014

Berechnungsmethode der Exemplarnummer

Das Obige ist die Berechnungsmethode zur Bestimmung der Menge.Dies ist ein mathematisches Problem, das nach dem Abschluss der Mittelschule gelöst werden kann, und mathematische Probleme werden im Allgemeinen durch Computer gelöst.Wenn Sie es nicht verstehen, können Sie zur Kommunikation kommen.
relative Quantifizierung
Die relative Quantifizierung wird hauptsächlich in der wissenschaftlichen Forschung eingesetzt.Wie viele Viren sind in 1 ml Blut enthalten und es handelt sich um ein DNA-Virus? Dies ist ein relativ deterministisches Ereignis: Die Blutmenge kann bestimmt werden und das DNA-Virus ist relativ stabil.Für uns ist es jedoch schwierig, die Anzahl der Transkriptionskopien eines bestimmten Gens in einem Blatt zu vergleichen, da es schwierig ist, die Größe, das Gewicht und die Zartheit des Blattes zu bestimmen, die Menge der extrahierten RNA schwer zu bestimmen ist und die Effizienz der umgekehrten Transkription ebenfalls schwer zu bestimmen ist. Das heißt, jeder Schritt kann dazu führen, dass die experimentellen Daten Fehler aufweisen und nicht verwendet werden können.
Daher muss die relative Quantifizierung ein Element einführen:das interne Referenzgen.
Mit anderen Worten: Bei der relativen Quantifizierung handelt es sich tatsächlich um einen Vergleich zwischen dem Zielgen und dem internen Referenzgen.Im Vergleich zum gleichen Gewebe und zur gleichen Zelle ist der Einfluss der Probengröße, der RNA-Extraktionsmenge, der Effizienz der reversen Transkription und der PCR-Effizienz relativ gering.Aufgrund der geringen Stichprobengröße waren sowohl interne Referenzgene als auch Zielgene relativ reduziert.Aus diesem Grund haben wir schon früher Wert auf Einheitlichkeit und Stabilität gelegt.
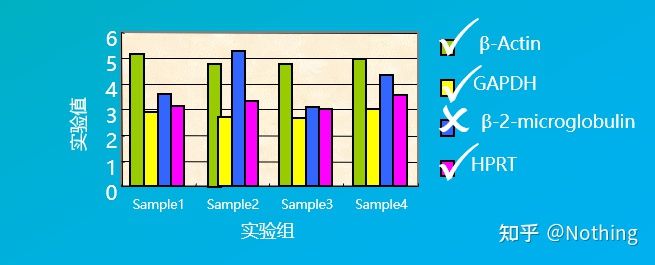
Interne Referenzgene sind im AllgemeinenHousekeeping-Gene(Haushaltsgene), die sich auf eine Klasse von Genen beziehen, die in allen Zellen stabil exprimiert werden, und deren Produkte notwendig sind, um die grundlegenden Lebensaktivitäten der Zellen aufrechtzuerhalten.
Verwechseln Sie dieses Konzept nicht.Bei Housekeeping-Genen handelt es sich um biologische Funktionsbegriffe, bei internen Referenzgenen um experimentelle Fachbegriffe.Housekeeping-Gene müssen eine Validierung bestehen, bevor sie als interne Referenzgene ausgewählt werden können.
Beispielsweise haben wir in der folgenden Abbildung mehrere Housekeeping-Gene ausgewählt, um deren Expressionsniveaus in verschiedenen Gewebezellen zu testen. Dabei haben wir festgestellt, dass sich die Expressionsniveaus von β-2-Mikroglobulin deutlich von denen der anderen drei Gene unterschieden, sodass sie nicht als interne Referenzgene verwendet werden konnten.
Nach dem Verständnis der Korrekturfunktion des internen Referenzgens werden aufgrund der Einführung des internen Referenzgens zwei Algorithmen abgeleitet.
·Doppelte Standardkurvenmethode
·2 – △△Ct-Methode (CT-Wert-Vergleichsmethode)
Wenn Sie daran interessiert sind, Arten und Genfunktionen zu untersuchen, geben Sie bitte die Forschung zu Algorithmen auf und verwenden Sie direkt Formeln oder Maschinen direkt.Wenn Sie ein heterosexueller Typ in Mathematik und Ingenieurwissenschaften sind, fühlen Sie sich bitte frei.
Doppelte Standardkurvenmethode
Quantifizieren Sie das Zielgen und das Housekeeping-Gen der Kontrollprobe und der zu testenden Probe anhand der Standardkurve und berechnen Sie dann den relativen Wert gemäß der Berechnungsformel, bei der es sich um das relative Expressionsniveau handelt.
Vorteile: einfache Analyse, relativ einfache experimentelle Optimierung
Nachteil: Für jedes Gen muss in jeder Versuchsrunde eine Standardkurve erstellt werden
Anwendung: Eine der beiden am häufigsten verwendeten und anerkanntesten relativen quantitativen Methoden bei der Untersuchung der Genexpressionsregulation
Die Formel lautet wie folgt:

Beispiele sind wie folgt:
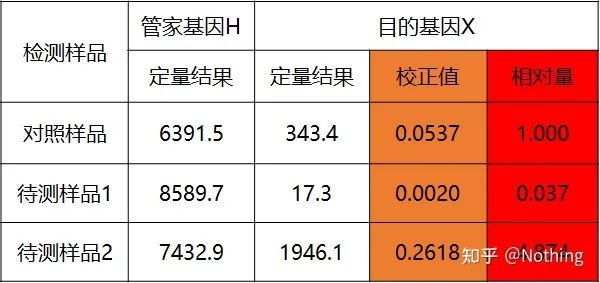
Berechnen Sie den relativen Betrag anhand des quantitativen Ergebnisses
2 – △△Ct-Methode (CT-Wert-Vergleichsmethode)

Vorteile: Es muss keine Standardkurve erstellt werden
Nachteile: Es wird davon ausgegangen, dass die Verstärkungseffizienz nahezu 100 % beträgt;die Standardabweichung beträgt < 5 % und es wird davon ausgegangen, dass die Standardkurve und die Effizienz zwischen den einzelnen Amplifikationen konsistent sind;die Optimierung der Versuchsbedingungen ist komplizierter.
Anwendung: Eine der beiden am häufigsten verwendeten und anerkanntesten relativen quantitativen Methoden bei der Untersuchung der Genexpressionsregulation
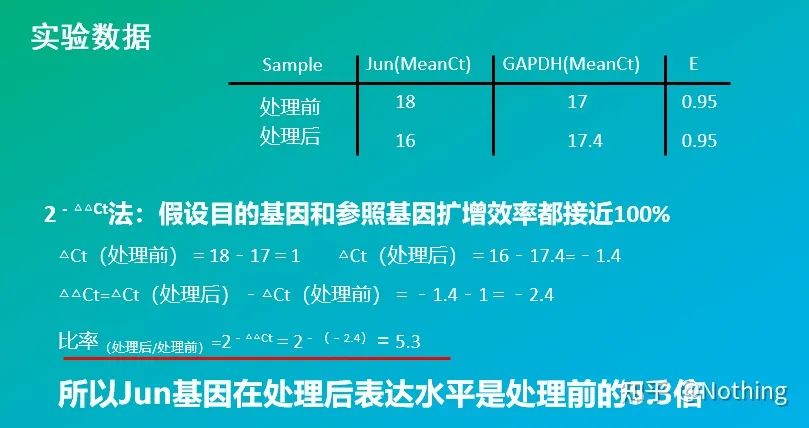
Natürlich kann die Amplifikationseffizienz normalerweise nicht perfekt sein. 1. Korrekturmethode: Wenn wir wissen, dass das Zielgen und das Referenzgen die gleiche Amplifikationseffizienz haben, die Amplifikationseffizienz jedoch nicht gleich 1 ist, kann 2-△△Ct wie folgt korrigiert werden: (1+E)-△△Ct. Wenn die Amplifikationseffizienz beispielsweise 0,95 beträgt, kann die Berechnungsformel auf 1,95 korrigiert werden -△△Ct
Bisher ist der Inhalt der quantitativen Fluoreszenz-PCR abgeschlossen.
Zeitpunkt der Veröffentlichung: 06.04.2023



